
Genetic Testing for Hereditary Hearing Loss - CAM 294
Description
Hearing Hearing loss is among the most etiologically heterogeneous disorders. More than 400 genetic syndromes include hearing loss as a feature; additionally, more than 100 genes are associated with nonsyndromic genetic hearing loss, and several non-genetic causes can also result in hearing loss. Genes associated with syndromic and nonsyndromic genetic hearing loss encode a variety of proteins involved in the development and function of the auditory system, including transcription factors, structural proteins, gap junction proteins, and ion channel.1 The genes may be associated with an autosomal dominant, autosomal recessive, X-linked, or mitochondrial inheritance pattern.2 Genetic counseling is strongly recommended for individuals pursuing genetic testing for nonsyndromic hereditary hearing loss.
For guidance on prenatal screening and preconception screening for hereditary hearing loss, please see CAM 358-Prenatal Screening (Genetic).
Regulatory Status
A search for “hearing loss” on the FDA website on March 29, 2021 did not yield any genetic results (FDA, 2021). Additionally, many labs have developed specific tests that they must validate and perform in house. These laboratory-developed tests (LDTs) are regulated by the Centers for Medicare & Medicaid Services (CMS) as high-complexity tests under the Clinical Laboratory Improvement Amendments of 1988 (CLIA ’88). As an LDT, the U.S. Food and Drug Administration has not approved or cleared this test; however, FDA clearance or approval is not currently required for clinical use.
Related Policies
70105 Cochlear Implant
204102 Whole Exome Sequencing
Policy
Application of coverage criteria is dependent upon an individual’s benefit coverage at the time of the request.
- For individuals who are in a family with a deleterious familial hearing loss gene mutation, the following genetic testing is considered MEDICALLY NECESSARY:
- Testing restricted to the known familial mutation
- Comprehensive genetic testing using multi-gene panel testing when the specific familial mutation is unknown
- For individuals diagnosed with hearing loss (when hearing loss due to nonhereditary causes [e.g., infection, injury, age-related] has been excluded), multi-gene panel testing (panel must include GJB2 and GJB6) is considered MEDICALLY NECESSARY.
The following does not meet coverage criteria due to a lack of available published scientific literature confirming that the test(s) is/are required and beneficial for the diagnosis and treatment of an individual’s illness.
- Repeat genetic testing for hereditary hearing loss-related mutations more than once per lifetime investigational and/or unproven and therefore considered NOT MEDICALLY NECESSARY.
- For all other situations not described above, genetic testing for hearing loss investigational and/or unproven and therefore considered NOT MEDICALLY NECESSARY.
NOTES:
Note: For two or more gene tests being run on the same platform, please refer to CAM 235 Laboratory Guideline Policy.
Table of Terminology
| Term |
Definition |
| AAO |
American Academy of Otolaryngology |
| AAP |
American Academy of Pediatrics |
| ACMG |
American College of Medical Genetics and Genomics |
| ACTG1 |
Actin gamma 1 |
| ANSD |
Auditory neuropathy spectrum disorder |
| ASHA |
American Speech-Language-Hearing Association |
| BSND |
Barttin CLCNK type accessory subunit beta |
| CDH23 |
Cadherin related 23 |
| CLDN14 |
Claudin 14 |
| CLIA ’88 |
Clinical Laboratory Improvement Amendments of 1988 |
| CLRN1 |
Clarin 1 |
| CMS |
Centers for Medicare & Medicaid |
| CMV |
Cytomegalovirus |
| CNV |
Contingent negative variation |
| COCH |
Cochlin |
| COL11A2 |
Collagen type Xl alpha 2 chain |
| DDPCR | Droplet digital polymerase chain reaction |
| DFN |
Different gene loci for nonsyndromic deafness |
| DFNA |
Autosomal dominant inherited deafness |
| DFNA5 |
Nonsyndromic hearing impairment protein 5 |
| DFNB |
Autosomal recessive inherited deafness |
| DFNB1 |
Nonsyndromic hearing loss and deafness |
| DFNB31 |
Deafness autosomal recessive 31 - Whirlin |
| DFNB59 |
Deafness autosomal recessive 59 |
| DFNX |
X-linked Inheritance |
| DNA |
Deoxyribonucleic acid |
| EKG |
Electrocardiogram |
| ESPN |
Espin |
| ESRRB |
Estrogen related receptor beta |
| EYA4 |
Transcriptional coactivator and phosphatase 4 |
| FDA |
Food And Drug Administration |
| GJB2 |
Gap junction protein, beta 2 |
| GJB6 |
Gap junction protein, beta 6 |
| GPR98 |
G-protein coupled receptor 98 |
| GRXCR1 |
Glutaredoxin and cysteine rich domain containing 1 |
| HGF |
Hepatocyte growth factor |
| HHL | Hereditary Hearing Loss |
| IPOG |
International Pediatric Otolaryngology Group |
| JCIH |
Joint Commission on Infant Hearing |
| KCNQ4 |
Potassium voltage-gated channel subfamily Q member 4 |
| LDTs |
Laboratory developed tests |
| LHFPL5 |
LHFPL tetraspan subfamily member 5 |
| MARVELD2 |
MARVEL domain containing 2 |
| MT-RNR1 |
Mitochondrially encoded 12S ribosomal ribonucleic acid |
| MT-TS1 |
Mitochondrially encoded transfer ribonucleic acid-SER |
| MYO15A |
Myosin XVA |
| MYO6 |
Myosin VI |
| MYO7A |
Myosin VIIA |
| NGS |
Next-generation sequencing |
| NSHL |
Nonsyndromic hearing loss |
| OMIM |
Online Mendelian Inheritance in Man |
| OTOA |
Otoancorin |
| OTOF |
Otoferlin |
| PCDH15 |
Protocadherin related 15 |
| PCR |
Polymerase chain reaction |
| PGT | Preimplantation genetic testing |
| POU3F4 |
POU class 3 homeobox 4 |
| PTPRQ |
Protein tyrosine phosphatase receptor type Q |
| RDX |
Radixin |
| SLC26A4 |
Solute carrier family 26 member 4 |
| SNL |
Sensorineural hearing loss |
| STRC |
Stereocilin |
| TECTA |
Tectorin alpha |
| TMC1 |
Transmembrane channel like 1 |
| TMIE |
Transmembrane inner ear |
| TMPRSS3 |
Transmembrane serine protease 3 |
| TRIOBP |
TRIO and F-actin binding protein |
| USH1C |
Usher syndrome type 1 protein component harmonin |
| WES |
Whole exome sequencing |
| WFS1 |
Wolframin ER transmembrane glycoprotein |
| WGS |
Whole genome sequencing |
| WHRN |
Whirlin |
Rationale
Approximately one in every 500 children born in the United States is deaf or has a hearing loss significant enough to affect speech and language development. Ninety-five percent of newborns with hearing loss identified by newborn hearing screening programs are born to hearing parents, obscuring the fact that most newborns have a hereditary cause for their hearing loss.1 Approximately 80 percent of cases of hereditary hearing loss are inherited in an autosomal recessive pattern, 19 percent are autosomal dominant, and the remaining cases X-linked (mainly recessive) or mitochondrial.2
Hearing loss is typically described in terms related to its clinical presentation. In general, it is categorized as either syndromic or nonsyndromic. Syndromic hearing loss is associated with other medical or physical findings, including malformations of the external ear or other organs, or with medical problems involving other organ systems. An estimated 30% of hereditary hearing loss is syndromic. Nonsyndromic hearing loss (NSHL) is defined as hearing loss that is not associated with visible abnormalities of the external ear or any related medical problems. For NSHL, it is more difficult to determine whether the etiology is hereditary or acquired because there are no other clinical manifestations at the time of the hearing loss presentation. NSHL accounts for an estimated 70% of genetically determined hearing loss,3 and it is frequently congenital and sensorineural.4
Increasing evidence implicates STRC as a major contributor to NSHL, with prevalence estimates approaching those of connexin-related hearing loss.5 Molecular diagnosis of STRC-associated hearing loss remains challenging due to the gene’s location within a tandemly duplicated region and the presence of a highly homologous pseudogene (STRCP1), which limits the sensitivity of standard next-generation sequencing (NGS) approaches. While the most common cause of DFNB16 is a homozygous contiguous gene deletion at 15q15.3, additional copy-number variants (CNVs) involving STRC are incompletely characterized, contributing to underdiagnosis and diagnostic uncertainty in affected families.5
The genetic loci where mutations associated with nonsyndromic hereditary hearing loss are commonly found are termed DFN. DFN loci are named based on their mode of inheritance: DFNA associated with autosomal dominant inheritance; DFNB with autosomal recessive inheritance; and DFNX with X-linked inheritance.2 The DFNB1 locus, which includes the GJB2 gene encoding the gap junction protein connexin 26 and the GJB6 gene encoding the gap junction protein connexin 30, accounts for an estimated 50% of all autosomal recessive nonsyndromic hearing loss and 15–40% of all deaf individuals in a variety of populations.1
GJB2 is a small gene with a single coding exon, which codes for the Cx26 connexin protein.6 At least 83 deafness-causing variants have been identified in GJB2, but a few common mutations account for a large percentage of alleles in several populations. Probands with this mutation generally have congenital hearing loss.2 Mutations in the GJB6 gene lead to similar effects on abnormal expression of connexin protein Cx30.7 GJB6 deletions have been observed in multiple populations, although they appear to be a relatively uncommon explanation for hearing loss in the United States.1
In addition to mutations in the GJB6 and GJB2 genes, many less common pathologic mutations are found in other genes. Some of these are: ACTG1, BSND, CDH23, CLDN14, COCH, COL11A2, DFNA5, DFNB31, DFNB59, ESPN, ESRRB, EYA4, GRXCR1, HGF, KCNQ4, LHFPL5, MARVELD2, MT-TS1, MYO15A, MYO6, MYO7A, OTOA, OTOF, PCDH15, POU3F4, PTPRQ, RDX, SLC26A4, STRC, TECTA, TMC1, TMIE, TMPRSS3, TRIOBP, USH1C, WFS1, and WHRN genes.2 Several gene panels exist for assessment of hereditary hearing loss. For example, Shang et al evaluated the “MiamiOtoGenes” panel, which consists of 180 genes. The investigators examined five unrelated probands with varying degrees of hearing loss onset and severity and found seven different genetic variants.8 Other entities offering proprietary genetic panels include BluePrint (239 genes), GeneDx (146 genes), OtoSCOPE by the University of Iowa (152 genes), GxVision® (245 genes), OtoGenomeTM Test (84 genes), Invitae Comprehensive Deaf Panel (203 genes), and AudioloGene Hereditary Hearing Loss Panel by Mayo Clinic Laboratories (160 genes).9-15
Clinical Utility and Validity
Shearer and Smith (2015) performed a meta-analysis focusing on the current genetic tests used to evaluate hearing loss. A total of 20 studies were included, containing 426 controls and 603 patients with idiopathic hearing loss. Several genetic panels such as OtoGenetics Deafness Test and OtoGenomeTM were used. Overall, the controls showed good sensitivity and specificity (over 99%), and the diagnostic rate was found to be 41% (with a range of 10%-83%). The authors concluded that “comprehensive genetic testing should form the cornerstone of a tiered approach to clinical evaluation of patients with hearing loss along with history, physical exam, and audiometry and can determine further testing that may be required, if any.”16
Sloan-Heggen, et al. (2016) performed parallel sequencing on 1119 “sequentially accrued” patients. A total of 440 (39%) of these patients were found to have a genetic etiology for hearing loss. Pathogenic variants were found in 49 genes, and various alterations such as missense variants (49% of the alterations), copy number variants (18%), insertions or deletions (13%), and nonsense variants (8%) were found. The authors noted the wide variety of the genetic spectrum of hearing loss.4
D'Aguillo, et al. (2019) examined the role of genetic screening as an adjunct to universal newborn hearing screening. The authors evaluated 16 studies and identified the rate of children that passed the universal newborn hearing screening but who also tested positive on a genetic screening. Of the 137895 infants included in the studies, pathogenic mutations were detected in 8.66% of them. Of this cohort, 545 infants passed the universal screening, but also tested positive on a genetic screening (1.4%).17
Costales, et al. (2020) studied the application of Otogenetics, a Next Generation Sequencing panel, in 27 patients diagnosed with sensorineural hearing loss (SNL) within a childhood hearing loss unit. A genetic diagnosis of SNL was made in 56% (15/27) of the patients whereas 44% (12/27) had pathogenic variants in genes associated with isolated SNL, syndromic SNL, and non-syndromic SNL. According to the authors, this study demonstrated that "it is possible to implement genetic diagnosis in the daily routine.”18
Yang, et al. (2021) developed a multiplex PCR sequencing assay to sequence the GJB2, SLC26A4, and MT-RNR1 genes and demonstrated that genetic screening can play an important role in newborn hearing screening. To validate the assay, 103 samples with known genotypes were analyzed using the multiplex PCR, which accurately identified all the variants with a 100% sensitivity and specificity. In the pilot study, 300 samples were analyzed and 12.3% were found to carry at least one pathogenic variant in the GJB2, SLC26A4, and MT-RNR1 genes. The study also revealed that pathogenic variants in the GJB2 gene had an 8% carrier rate in the newborn population. The authors concluded that "the assay is an accurate and reliable test and can be used to screen genetic hearing loss in newborns.”19
In a cohort of 72 patients with DFNB16, Alvaro, et al. (2025) found that use of droplet digital PCR (ddPCR) improved detection and characterization of STRC copy number variants that are difficult to resolve using conventional next-generation sequencing, including complex rearrangements within highly homologous regions. This approach enabled more precise molecular diagnoses in children with mild-to-moderate hearing loss and identified male patients at risk for deafness–infertility syndrome due to involvement of the CATSPER2 gene. These findings suggest that ddPCR may enhance the clinical validity of genetic testing for suspected STRC-related hearing loss when used as a complementary method in selected cases.5 However, the evidence is limited by cohort size, lack of longitudinal outcome data, and the novel nature of some reported genomic associations, which remain unvalidated. Consequently, the clinical utility of ddPCR-based testing is currently confined to targeted diagnostic clarification and genetic counseling rather than routine first-line testing or population screening.
García-García, et al. (2020) analyzed 128 patients from 118 families with syndromic or nonsyndromic hearing loss (HL) using targeted NGS of 59 related genes. Variants were prioritized based on allele frequency and classified per American College of Medical Genetics and Genomics guidelines. Disease-causing variants were identified in 40% of families, spanning autosomal recessive (AR), autosomal dominant (AD), and X-linked inheritance patterns. Pathogenic or likely pathogenic variants were found in 26 genes (15 AR, 9 AD, and 2 X-linked), including 14 novel variants. The study emphasizes the clinical value of targeted NGS for diagnosing sensorineural HL and suggests that optimal gene panels should be tailored to population-specific genetic profiles and laboratory resources.20
Wang, et al. (2025) evaluated the clinical application of preimplantation genetic testing (PGT) for hereditary hearing loss (HHL) in high-risk families. Using low-coverage NGS combined with linkage analysis, the investigators demonstrated reliable identification and inheritance tracking of known pathogenic variants in embryos from 19 couples with established HHL-associated genotypes. A high proportion of embryos yielded interpretable results, enabling selection against transmission of deafness-related variants and resulting in unaffected live births following assisted reproduction.21 These findings support the clinical utility of genetic testing for HHL in the context of reproductive decision-making when integrated with genetic counseling and in vitro fertilization. However, the evidence is limited by a small, highly selected cohort, reliance on previously characterized familial variants, and absence of population-based performance metrics; consequently, these results do not establish clinical validity or utility for population screening, novel variant detection, or routine use outside specialized reproductive settings.
Liao, et al. (2022) put together a retrospective cohort study of 2075 patients who were seen at the Children’s Communications Clinic; 517 patients completed a hearing loss testing with a GeneDx panel. Researchers gathered sociodemographic characteristics, hearing loss characteristics, and medical variables of hearing loss. On univariable analysis, those who received a genetic diagnosis were younger than those who did not receive a genetic diagnosis (OR, 0.92; 95% CI, 0.88-0.96). Twenty-four diagnostic genes were found in the study cohort, the most common diagnostic gene being GJB2, which encodes for connexin 26, a major protein found in gap junction protein in the cochlea. Additionally, the most prevalent diagnostic variant was different across races and ethnicities, that is, Asian patients were more likely to have the C.109 G>A variant and white patients were more likely to have the C.35del, and Hispanic patients were “almost equally” likely to have the c.35del variant as any other variant (aside from c.109 G>A).22
The American College of Medical Genetics and Genomics (ACMG)
In 2014, the ACMG issued the following guidelines for the clinical evaluation and diagnosis of hearing loss. For individuals lacking physical findings suggestive of a known syndrome and having medical and birth histories that do not suggest an environmental cause of hearing loss, ACMG recommends that a tiered diagnostic approach should be implemented.
- “Pretest genetic counseling should be provided, and, with patient’s informed consent, genetic testing should be ordered.”
- “Single-gene testing may be warranted in cases in which the medical or family history, or presentation of the hearing loss, suggests a specific etiology. For example, testing for mitochondrial DNA mutations associated with aminoglycoside ototoxicity may be considered for individuals with a history of use of aminoglycoside antibiotics.”
- “In the absence of any specific clinical indications and for singleton cases and cases with apparent autosomal recessive inheritance, the next step should be testing for DFNB1-related hearing loss (due to mutations in GJB2 and adjacent deletions in GJB6).”
- “If initial genetic testing is negative, genetic testing using gene panel tests, NGS technologies such as large sequencing panels targeted toward hearing loss-related genes, WES, or WGS may be considered. Because several tests are clinically available, the clinician must be aware of the genes included in the test (panel) chosen and the performance characteristics of the platform chosen, including coverage, analytic sensitivity, and what types of mutations will be detected. It should be noted that the cost of these new genetic sequencing technologies is decreasing so rapidly that a tiered approach to testing may soon no longer be cost effective. In particular, for large sequencing panels targeted toward hearing loss– related genes, it may, in some cases, already be more cost effective to use NGS technologies as the initial test in the evaluation of hearing loss. However, issues related to genomic testing, such as the likelihood of incidental findings, will have to be addressed.”
- “If genetic testing reveals mutation(s) in a hearing loss-related gene, mutation-specific genetic counseling should be provided, followed by appropriate medical evaluations and referrals.”
- “If genetic testing fails to identify an etiology for a patient’s hearing loss, the possibility of a genetic or acquired etiology remains. This point must be emphasized because it can be misunderstood by clinicians and by patients and their families. For interested patients and families, further genetic testing may be pursued on a research basis.”
- “CMV testing should be done at the same time as genetic testing for infants with congenital hearing loss. For later-onset or progressive hearing loss, CMV testing can be obtained, but the likelihood that a positive test is due to postnatal exposure increases with age.”
For individuals with findings that suggest a syndromic genetic etiology for their hearing loss:
- “Pretest genetic counseling should be provided, and, with patient’s informed consent, genetic testing, if available, should be ordered to confirm the diagnosis—this testing may include single-gene tests, hearing loss sequencing panels, WES, WGS, chromosome analysis, or microarray-based copy-number analysis, depending on clinical findings.”
- “Appropriate studies should be undertaken to determine whether other organs are involved; and
- “Appropriate near-term and long-term screening and management should be arranged, including referrals to specialists, as indicated by the associated manifestations of the particular syndrome.”1
The ACMG also published an algorithm stating to “consider” GJB2, GJB6 or other gene specific testing if familial or nonsyndromic hearing loss was suspected. If nonsyndromic and mitochondrial inheritance was suspected, the ACMG recommended testing for the A1555G mutation.23
In 2021, ACMG released an updated guideline for screening for AR and X-linked conditions during pregnancy and preconception. Their practice resource aims to recommend “a consistent and equitable approach for offering carrier screening to all individuals during pregnancy and preconception” and replaces any earlier ACMG position statements on prenatal/preconception expanded carrier screening and provide the following recommendations:
- “Carrier screening enables those screened to consider their reproductive risks, reproductive options, and to make informed decisions.”
- “The phrase “expanded carrier screening” be replaced by “carrier screening.”
- “Adopting a more precise tiered system based on carrier frequency:
- Tier 4: <1/200 carrier frequency (includes Tier 3) genes/condition will vary by lab
- Tier 3: ≥ 1/200 carrier frequency (includes Tier 2) includes X-linked conditions
- Tier 2: ≥1/100 carrier frequency (includes Tier 1)
- Tier 1: CF [Cystic Fibrosis] + SMA [spinal muscular atrophy] + Risk Based Screening”
- “Tier 1 screening conveys the recommendations previously adopted by ACMG and ACOG” and “adopts an ethnic and population neutral approach when screening for cystic fibrosis and spinal muscular atrophy. Beyond these two conditions, additional carrier screening is determined after risk assessment, which incorporates personal medical and family history as well as laboratory and imaging information where appropriate”
- “Tier 2 carrier screening stems from an ACOG recommendation for conditions that have a severe or moderate phenotype and a carrier frequency of at least 1/100.” However, “data demonstrate that carrier screening for two common conditions using a carrier frequency threshold of 1/100 may not be equitable across diverse populations. Others have shown that limiting the carrier frequency to ≥1/100 creates missed opportunities to identify couples at risk for serious conditions.”
- “We define Tier 3 screening as carrier screening for conditions with a carrier frequency ≥1/200 . . . Tier 2 and Tier 3 screening prioritize carrier frequency as a way to think about conditions most appropriate for screening in the general population. However, when ACOG proposed this level, they did not specify whether it was thinking about carrier frequency in terms of the global population or subpopulations. We use “carrier frequency” to mean in any ethnic group with reasonable representation in the United States.”
- “Tier 4 includes genes less common than those in Tier 3 and can identify additional at-risk couples. Tier 4 has no lower limit carrier screening frequency and can greatly extend the number of conditions screened . . . the clinical validity at this level of carrier screening may be less compelling, therefore we suggest reserving this level of screening for consanguineous pregnancies (second cousins or closer) and in couples where family or medical history suggests Tier 4 screening might be beneficial . . . Importantly, patients should understand that their chance of being a carrier for one or more conditions increases as the number of conditions screened is increased.”
- “All pregnant patients and those planning a pregnancy should be offered Tier 3 carrier screening.
- Tier 4 screening should be considered:
- When a pregnancy stems from a known or possible consanguineous relationship (second cousins or closer);
- When a family or personal medical history warrants.
- ACMG does NOT recommend:
- Offering Tier 1 and/or Tier 2 screening, because these do not provide equitable evaluation of all racial/ethnic groups.
- Routine offering of Tier 4 panels.”
- “Carrier screening paradigms should be ethnic and population neutral and more inclusive of diverse populations to promote equity and inclusion.”
- “All pregnant patients and those planning a pregnancy should be offered Tier 3 carrier screening for autosomal recessive [Table 1] . . . conditions.”
- “Reproductive partners of pregnant patients and those planning a pregnancy may be offered Tier 3 carrier screening for autosomal recessive conditions [Table 1] when carrier screening is performed simultaneously with their partner.”
- “When Tier 1 or Tier 2 carrier screening was performed in a prior pregnancy, Tier 3 screening should be offered.”24
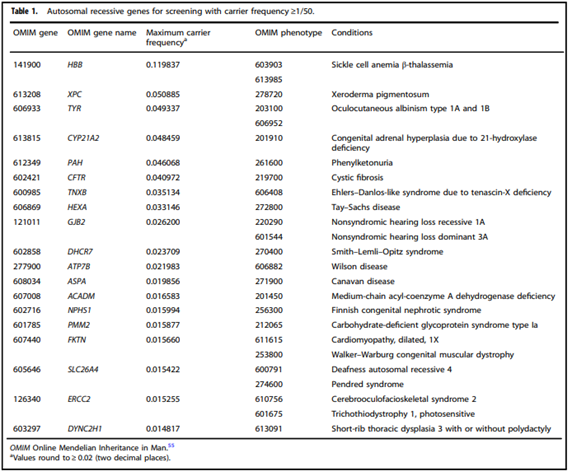
Gregg, et al. (2021)
In 2022, the ACMG released updated guidelines for clinical evaluation and etiologic diagnosis of hearing loss. The updated guideline suggests the following recommendations:
- “All newborns and infants with confirmed HL should undergo a comprehensive evaluation in which patient-focused medical and birth histories, a 3-generation pedigree, and family medical history are obtained, and a physical examination that focuses on dysmorphic physical findings is performed. Evaluation of children and young adults with HL should follow a similar approach. Evaluation of deaf or hard-of-hearing adults should be customized based on the age of onset and other characteristics of HL.”
- “The medical and birth histories may be helpful in differentiating between acquired vs inherited causes of HL.”
- “For individuals with findings that suggest a syndromic genetic etiology for their HL:
- “Pretest genetic counseling should be provided, and, with patient’s or caregiver’s informed consent, genetic testing should be ordered to confirm the diagnosis. This testing may include single-gene tests, HL multigene panels, ES, GS, chromosome analysis, or microarray- or NGS-based copy number analysis, depending on clinical findings.”
- “Appropriate studies should be undertaken to determine whether other organs are involved.”
- “Appropriate near-term and long-term screening and management should be arranged, including referrals to specialists, as indicated by the associated manifestations of the syndrome.”
- “For individuals lacking physical findings suggestive of a known syndrome, a tiered diagnostic approach should be implemented.”
- “Unless clinical and/or family history suggests a specific genetic etiology, comprehensive HL gene panel testing should be initiated. If panel testing is negative, genome-wide testing, such as ES or GS, may be considered.
- The HL panel should include the genes recommended by the HL Gene Curation Expert Panel [https://search.clinicalgenome.org/kb/affiliate/10007?page=1&size=25&search=].
- If genetic testing reveals variant(s) in an HL-related gene, gene-specific genetic counseling should be provided, followed by appropriate medical evaluations and referrals.
- If genetic testing fails to identify an etiology fora patient’s HL, the possibility of a genetic etiology remains.”
- “Temporal bone imaging by computed tomography or magnetic resonance imaging should be considered as a complement to genetic testing, particularly if the diagnosis remains unclear; if cochlear im-plantation is being considered; if auditory neuropathy is noted, in cases of progressive HL; or if other clinical concerns exist. The anticipated clinical utility of imaging studies should be balanced against the risks associated with radiation exposure and sedation.”
- "CMV testing should be done as soon as possible after birth but within the first 3 weeks of life for infants with congenital HL. For later-onset or progressive HL, CMV testing can be obtained, but the likelihood that a positive test is caused by postnatal exposure increases with age.”
- “Unless clinical and/or family history suggests a specific genetic etiology, comprehensive HL gene panel testing should be initiated. If panel testing is negative, genome-wide testing, such as ES or GS, may be considered.
- “Referral to a multidisciplinary care center, when available, is recommended.”
- “A team approach that includes otolaryngologists, clinical geneticists, genetic counselors, audiologists, speech and language specialists, early hearing intervention and family support specialists and other appropriate specialists offers optimal opportunity to provide ongoing management and support of deaf and hard-of-hearing individuals and their families as their needs change over time.”
- “For cases in which the genetic evaluation failed to identify an underlying cause, periodic follow-up care every 3 years with a geneticist may be appropriate for several reasons. First, subtle features of syndromic forms of HL may not be apparent at birth or early in childhood but may appear as deaf or hard-of-hearing individuals grow into adulthood. These may prompt additional medical tests or referrals for specialty care. Second, follow-up visits offer the opportunity to inform individuals about new genetic tests that may have become available or changes in the interpretation of previous test results as medical knowledge advances. Finally, follow-up visits may also help identify clinical concerns unrelated to HL, for which referral for specialty care may be appropriate.”
Regardless of whether genetic test results are positive, negative, or inconclusive, results should be communicated through the process of genetic counseling and potential risks to other family members should be conveyed.”25
The Joint Committee on Infant Hearing (JCIH)
In 2007, the JCIH recommended that evaluation of infants with confirmed hearing loss should include a review of family history of specific genetic disorders or syndromes, including genetic testing for gene mutations such as GJB2 (connexin-26), and syndromes commonly associated with early-onset childhood sensorineural hearing loss.26 In 2013, a supplement by the ASHA was added to the JCIH. The 2013 supplement also stated that medical providers must “understand atypical development etiologies and diagnoses, and refer for medical-genetic evaluation” and that families must be educated on the “importance of medical, genetic, ophthalmologic, and cardiac (EKG) evaluations on children with any type and degree of hearing loss.”27
In 2019, the JCIH published an updated position statement. They note that the ACMG recommends offering genetic counseling and testing to all infants who are deaf or hard-of-hearing and their families. A geneticist’s evaluation should include “a review of family history of specific genetic disorders or syndromes, genetic testing for gene mutations such as GJB2 (connexin-26), and syndromes commonly associated with early-onset hearing loss.”28
References
- Alford RL, Arnos KS, Fox M, et al. American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genetics in medicine : official journal of the American College of Medical Genetics. Apr 2014;16(4):347-55. doi:10.1038/gim.2014.2
- A Eliot Shearer MSH, and Richard JH Smith. Hereditary Hearing Loss and Deafness Overview. 2023.
- Angeli S, Lin X, Liu XZ. Genetics of hearing and deafness. Anatomical record (Hoboken, NJ : 2007). Nov 2012;295(11):1812-29. doi:10.1002/ar.22579
- Sloan-Heggen CM, Bierer AO, Shearer AE, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Human genetics. Apr 2016;135(4):441-50. doi:10.1007/s00439-016-1648-8
- Alvaro S, Castillo D, Genovés J, et al. Refining the detection of complex rearrangements in 15q15.3 region involving the STRC gene in hereditary hearing loss patients. Journal of Human Genetics. 2025/08/01 2025;70(8):395-403. doi:10.1038/s10038-025-01347-9
- OMIM. Gap Junction Protein Beta-2; GJB2. https://www.omim.org/entry/121011
- OMIM. Gap Junction Protein, Beta-6; GJB6. https://www.omim.org/entry/604418
- Shang H, Yan D, Tayebi N, et al. Targeted Next-Generation Sequencing of a Deafness Gene Panel (MiamiOtoGenes) Analysis in Families Unsuitable for Linkage Analysis. BioMed research international. 2018;2018:3103986. doi:10.1155/2018/3103986
- BluePrint. Comprehensive Hearing Loss and Deafness Panel. Updated January 17, 2025. https://blueprintgenetics.com/tests/panels/ear-nose-throat/comprehensive-hearing-loss-and-deafness-panel/
- GeneDx. Hearing Loss Panel. https://providers.genedx.com/tests/detail/hearing-loss-panel-925
- University of Iowa. OtoSCOPE Genetic Hearing Loss Testing v9. https://morl.lab.uiowa.edu/clinical-diagnostic-services/hearing-loss-clinical-division/otoscoper-genetic-hearing-loss-testing#accordion-item-10286-0
- Otogenetics. Clinical Genetic Testing and Research. https://www.otogenetics.com/
- Partners Healthcare. OtoGenome™ Test for Hearing Loss and Related Syndromes (110 Genes) Details. https://personalizedmedicine.partners.org/Laboratory-For-Molecular-Medicine/Tests/Hearing-Loss/OtoGenome.aspx
- Invitae. Invitae Comprehensive Deafness Panel. https://www.invitae.com/us/providers/test-catalog/test-55009
- Mayo Clinic. Hearing Loss Panel. https://www.mayocliniclabs.com/test-catalog/overview/619372
- Shearer AE, Smith RJ. Massively Parallel Sequencing for Genetic Diagnosis of Hearing Loss: The New Standard of Care. Otolaryngology--head and neck surgery : official journal of American Academy of Otolaryngology-Head and Neck Surgery. Aug 2015;153(2):175-82. doi:10.1177/0194599815591156
- D'Aguillo C, Bressler S, Yan D, et al. Genetic screening as an adjunct to universal newborn hearing screening: literature review and implications for non-congenital pre-lingual hearing loss. 2019;(1708-8186 (Electronic))
- Costales M, Diñeiro M, Cifuentes GÁ, et al. Clinical Utility of Next-generation Sequencing in the Aetiological Diagnosis of Sensorineural Hearing Loss in a Childhood Hearing Loss Unit. Acta Otorrinolaringologica (English Edition). 2020/05/01/ 2020;71(3):166-174. doi:10.1016/j.otoeng.2019.05.005
- Yang H, Luo H, Zhang G, Zhang J, Peng Z, Xiang J. A multiplex PCR amplicon sequencing assay to screen genetic hearing loss variants in newborns. BMC Medical Genomics. 2021/02/27 2021;14(1):61. doi:10.1186/s12920-021-00906-1
- García-García G, Berzal-Serrano A, García-Díaz P, et al. Improving the Management of Patients with Hearing Loss by the Implementation of an NGS Panel in Clinical Practice. Genes (Basel). Dec 7 2020;11(12)doi:10.3390/genes11121467
- Wang W, Guan J, Ma M, et al. Clinical application of preimplantation genetic testing based on low-coverage next-generation sequencing with linkage analyses in hereditary hearing loss families. Journal of Assisted Reproduction and Genetics. 2025/06/01 2025;42(6):1989-2002. doi:10.1007/s10815-025-03504-7
- Liao EN, Taketa E, Mohamad NI, Chan DK. Outcomes of Gene Panel Testing for Sensorineural Hearing Loss in a Diverse Patient Cohort. JAMA Network Open. 2022;5(9):e2233441-e2233441. doi:10.1001/jamanetworkopen.2022.33441
- ACMG. Hearing Loss https://www.acmg.net/PDFLibrary/Hearing-Loss-Algorithm.pdf
- Gregg AR, Aarabi M, Klugman S, et al. Screening for autosomal recessive and X-linked conditions during pregnancy and preconception: a practice resource of the American College of Medical Genetics and Genomics (ACMG). Genetics in medicine : official journal of the American College of Medical Genetics. Oct 2021;23(10):1793-1806. doi:10.1038/s41436-021-01203-z
- Li MM, Tayoun AA, Distefano M, et al. Clinical evaluation and etiologic diagnosis of hearing loss: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine. 2022;24(7):1392-1406. doi:10.1016/j.gim.2022.03.018
- JCIH. Year 2007 Position Statement: Principles and Guidelines for Early Hearing Detection and Intervention Programs. Pediatrics. 2007;120(4):898. doi:10.1542/peds.2007-2333
- ASHA. Supplement to the JCIH 2007 position statement: principles and guidelines for early intervention following confirmation that a child is deaf or hard of hearing [Position Statement]. American Speech-Language-Hearing Association. https://www.asha.org/policy/ps2013-00339/
- JCIH. Year 2019 Position Statement: Principles and Guidelines for Early Hearing Detection and Intervention Programs. Journal of Early Hearing Detection and Intervention. 2019;4(2):1-44. doi:10.15142/fptk-b748
Coding Section
| Codes | Number | Description |
| CPT | 81253 | GJB2 (gap junction protein, beta 2, 26kDa, connexin 26) (e.g., nonsyndromic hearing loss) gene analysis; known familial variants |
| 81403 | Molecular pathology procedure, Level 4 (eg, analysis of single exon by DNA sequence analysis, analysis of >10 amplicons using multiplex PCR in 2 or more independent reactions, mutation scanning or duplication/deletion variants of 2-5 exons | |
| 81430 | Hearing loss (e.g., nonsyndromic hearing loss, Usher syndrome, Pendred syndrome); genomic sequence analysis panel, must include sequencing of at least 60 genes, including CDH23, CLRN1, GJB2, GPR98, MTRNR1, MYO7A, MYO15A, PCDH15, OTOF, SLC26A4, TMC1, TMPRSS3, USH1C, USH1G, USH2A, and WFS1 | |
| 81431 | Hearing loss (e.g., nonsyndromic hearing loss, Usher syndrome, Pendred syndrome); duplication/deletion analysis panel, must include copy number analyses for STRC and DFNB1 deletions in GJB2 and GJB6 genes | |
| 81479 | Unlisted molecular pathology procedure | |
| 96040 | Medical genetics and genetic counseling services, each 30 minutes face-to-face with patient/family | |
| S0265 | Genetic counseling under physician supervision, each 15 minutes | |
| ICD-10-CM (effective 10/01/15) | H90.0-H91.8X9 | Conductive and sensorineural hearing loss code range |
| H90.3 | Sensorineural hearing loss, bilateral | |
| H90.41 | Sensorineural hearing loss, unilateral, right ear, with unrestricted hearing on the contralateral side | |
| H90.42 | Sensorineural hearing loss, unilateral, left ear, with unrestricted hearing on the contralateral side | |
| H90.5 | Unspecified sensorineural hearing loss | |
| H90.6 | Mixed conductive and sensorineural hearing loss, bilateral | |
| H90.71 | Mixed conductive and sensorineural hearing loss, unilateral, right ear, with unrestricted hearing on the contralateral side | |
| H90.72 | Mixed conductive and sensorineural hearing loss, unilateral, left ear, with unrestricted hearing on the contralateral side | |
| H91.8X1 | Other specified hearing loss, right ear | |
| H91.8X2 | Other specified hearing loss, left ear | |
| H91.8X3 | Other specified hearing loss, bilateral | |
| Z13.71 | Encounter for nonprocreative screening for Genetic Disease carrier status | |
| Z14.8 | Genetic carrier of other disease | |
| Z15.89 | Genetic susceptibility to other disease | |
| Z31.430 | Encounter of female for testing for genetic disease carrier status for procreative management | |
| Z31.440 | Encounter of male for testing for genetic disease carrier status for procreative management | |
| Z82.2 | Family history of deafness and hearing loss | |
| ICD-10-PCS (effective 10/01/15) | Not applicable. ICD-10-PCS codes are only used for inpatient services. There are no ICD procedure codes for laboratory test. | |
| Type of Service | ||
| Place of Service |
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community and other nonaffiliated technology evaluation centers, reference to federal regulations, other plan medical policies, and accredited national guidelines.
"Current Procedural Terminology © American Medical Association. All Rights Reserved"
History From 2014 Forward
| 05/06/2026 | Annual review, no change to policy intent. Updating table of terminology, rationale, and references. |
| 07/23/2025 | Annual review, updating coverage criteria 3 for repeat genetic testing more than once per lifetime is not medically necessary. Also updating description, note, rationale, references. Removing CPT 81252, 81254, S3844; adding CPT 81403 and 81479. |
| 08/05/2024 | Annual review, updating rationale and references and updating note #2 to direct reader to CAM 235. |
| 07/28/2023 | Annual review, updating for clarity and consistency. Updating criteria to mirror recommendations from ACMG. Also updating description, table of terminology, rational and references. |
| 08/08/2022 | Annual review, no change to policy intent. Updating coding. |
| 07/13/2021 |
Annual review, no change to policy intent. Updating description, rationale, references and coding. |
| 07/02/2020 |
Annual review, no change to policy intent. Updating coding. |
| 07/12/2019 |
Annual review, updating title to remove "nonsyndromic", updating verbiage in criteria for implementation and clarity, adding verbiage regarding gene panel tests/ NGS technologies for suspected syndromic hearing loss. |
| 06/19/2019 |
Interim review. Genetic counseling is recommended is replacing Genetic counseling is Medically necessary. No other changes made. |
| 07/26/2018 |
Annual review, adding coverage statement regarding genetic counseling, reformatting some policy language for clarity. Updating coding. |
| 06/21/2017 |
Interim review, updating title, background, description, references, rationale and policy. |
| 04/25/2017 |
Updated category to Laboratory. No other changes. |
| 12/05/2016 |
Annual review, no change to policy intent. |
| 11/12/2015 |
Annual review, no change to policy intent. Updated background, description, regulatory status, guidelines, rationale and references. Added appendix 1. |
| 12/02/2014 |
New Policy |