
Genomic Testing for Hematopoietic Neoplasms - CAM 390
Description
Hematologic cancers impact the normal production and function of blood cells. These cancers often begin in bone marrow, where stem cells develop into white blood cells, red blood cells, or platelets. Hematologic cancers such as myeloid and lymphoid neoplasms occur when there is an uncontrolled growth of abnormal cells which overtake the development of normal blood cells. An overabundance of abnormal cells interferes with the regular functions of normal cells.1 Myeloid neoplasms occur when there is a proliferation of myeloid cells that have originated from a primitive stem cell.2 In contrast, lymphoid neoplasms are composed of a lymphocytic cell population which is usually malignant (clonal) by molecular genetic and/or immunophenotypic analysis.3
For guidance on flow cytometry in hematopoietic neoplasms, please refer to CAM 120 Flow Cytometry. For guidance on minimal/measurable residual disease (MRD) in hematopoietic neoplasms, please refer to CAM 251 Minimal Residual Disease.
Regulatory Status
On April 28, 2017 the FDA approved the LeukoStrat® CDx FLT3 Mutation Assay as a “PCR-based, in vitro diagnostic test designed to detect internal tandem duplication (ITD) mutations and the tyrosine kinase domain mutations D835 and I836 in the FLT3 gene in genomic DNA extracted from mononuclear cells obtained from peripheral blood or bone marrow aspirates of patients diagnosed with acute myelogenous leukemia (AML). The LeukoStrat® CDx FLT3 Mutation Assay is used as an aid in the selection of patients with AML for whom RYDAPT® (midostaurin) treatment is being considered. The Leukostrat® CDx FLT3 Mutation Assay is to be performed only at Laboratory for Personalized Molecular Medicine (LabPMM) LLC, a single site laboratory located at 6330 Nancy Ridge Dr., San Diego, CA 92121.”
On Aug. 1, 2017 the FDA approved the Abbott RealTime IDH2 as an “in vitro polymerase chain reaction (PCR) assay for the qualitative detection of single nucleotide variants (SNVs) coding nine IDH2 mutations (R140Q, R140L, R140G, R140W, R172K, R172M, R172G, R172S, and R172W) in DNA extracted from human blood (EDTA) or bone marrow (EDTA). Abbott RealTime IDH2 is for use with the Abbott m2000rt System. Abbott RealTime IDH2 is indicated as an aid in identifying acute myeloid leukemia (AML) patients with an isocitrate dehydrogenase-2 (IDH2) mutation for treatment with IDHIFA® (enasidenib).”
Many labs have developed specific tests that they must validate and perform in house. These laboratory-developed tests (LDTs) are regulated by the Centers for Medicare & Medicaid Services (CMS) as high-complexity tests under the Clinical Laboratory Improvement Amendments of 1988 (CLIA ’88). LDTs are not approved or cleared by the U.S. Food and Drug Administration; however, FDA clearance or approval is not currently required for clinical use.
Policy
Application of coverage criteria is dependent upon an individual’s benefit coverage at the time of the request.
- For the initial diagnosis and profiling of hematopoietic neoplasms, the following testing is considered MEDICALLY NECESSARY:
- Bone marrow cytogenetics and/or fluorescent in situ hybridization (FISH) of bone marrow samples.
- Multigene panel testing.
- For the differential diagnosis of chronic myeloid leukemia (CML) or acute lymphoblastic leukemia (ALL) or for optimal risk stratification and treatment planning for individuals diagnosed with B-cell ALL (B-ALL), one time qualitative or quantitative RT-PCR testing on blood or bone marrow for identification of the BCR-ABL1 fusion gene transcript type, including determination of transcript size (e.g., p190 vs. p210), is considered MEDICALLY NECESSARY.
- For individuals diagnosed with acute lymphoblastic leukemia (ALL) or pediatric ALL (PEDALL) who are under surveillance, the following tests is considered MEDICALLY NECESSARY once every 3 to 6 months for at least 5 years:
- Bone marrow cytogenetics.
- Bone marrow FISH.
- Multigene panel testing.
- For all individuals with Philadelphia chromosome positive ALL (Ph+ ALL), BCR-ABL1 transcript-specific quantification is considered MEDICALLY NECESSARY at the following frequency:
- Once per month for individuals who have detectable levels of BCR-ABL1 transcript.
- Once every three months for individuals who have undetectable levels of BCR-ABL1 transcript.
- For individuals diagnosed with acute myeloid leukemia (AML) who had cytogenetic abnormalities at diagnosis, cytogenetic or FISH testing is considered MEDICALLY NECESSARY at the following time points:
- Fourteen to twenty-one days after the start of therapy to document hypoplasia.
- When hypoplasia is documented, repeat analysis seven to fourteen days later to clarify persistence of leukemia.
- When hypoplasia is not documented, repeat analysis at the time of hematologic recovery to document remission.
- By 42 days post-treatment with either standard-dose or high-dose cytarabine induction for poor-risk AML, regardless of the degree of hematologic recovery.
- Fourteen to twenty-one days after the start of therapy to document hypoplasia.
- For individuals with AML who have experienced relapsed/refractory disease after the completion of post-remission therapy, multigene panel testing is considered MEDICALLY NECESSARY.
- For optimal risk stratification and treatment planning for individuals diagnosed with chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL), one time testing with any of the following tests (when not already performed during initial diagnosis) is considered MEDICALLY NECESSARY:
- FISH to detect +12; del(11q); del(13q); del(17p).
- TP53 mutation testing.
- CpG-stimulated metaphase karyotype for complex karyotype.
- Immunoglobulin heavy chain variable region gene (IGHV) mutation testing.
- For individuals diagnosed with chronic myeloid leukemia (CML), quantitative reverse transcription polymerase chain reaction (qPCR) testing for the BCR-ABL1 fusion gene transcript is considered MEDICALLY NECESSARY at the following frequency:
- To establish baseline levels at diagnosis.
- For individuals undergoing TKI therapy:
- Every 3 months after initiation of therapy until major molecular response (MMR) (BCR-ABL1 [IS] < 1% [> 0.1% – 1%]) has been achieved.
- Every 3 months for 2 years and every 3 – 6 months thereafter.
- If there is a 1-log increase in BCR-ABL1 transcript levels with MMR, repeat in 1 – 3 months.
- For individuals diagnosed with CML who are pursuing or who are actively undergoing TKI therapy, bone marrow cytogenetics is considered MEDICALLY NECESSARY at the following time points:
- To establish cytogenetic changes.
- When there is a failure to reach response milestones.
- When there is any sign of loss of hematologic response.
- When there is any sign of loss of complete cytogenetic response (CCyR) or its molecular response correlate, defined as an increase in BCR-ABL1 transcript to >1%.
- Evaluation of BCR-ABL kinase domain point mutations in patients with CML is considered MEDICALLY NECESSARY at the following time points:
- When the individual has chronic phase CML.
- When there is failure to reach response milestones.
- When there is any sign of loss of hematologic response.
- When there is any sign of loss of CCyR or its molecular response correlate, defined as an increase in BCR-ABL1 transcript to 1%.
- When there is a 1-log increase in BCR-ABL1 transcript levels and a loss of MMR.
- When the disease progresses to accelerated or blast phase.
- For individuals with CML who are undergoing treatment discontinuation with TKI therapy and who remain in MMR after discontinuation of therapy, qPCR on blood or bone marrow for the BCR-ABL1 fusion gene transcription is considered MEDICALLY NECESSARY at the following frequency:
- Every month for the first 6 months following discontinuation.
- Once every two months in months 7-12 following discontinuation.
- Once every three months beginning 12 months following discontinuation of therapy, as long as the individual remains in MMR.
- For individuals with CML, the use of FISH to monitor response to TKI therapy is considered NOT MEDICALLY NECESSARY.
- Simultaneous testing of both bone marrow and blood for monitoring purposes is considered NOT MEDICALLY NECESSARY.
The following does not meet coverage criteria due to a lack of available published scientific literature confirming that the test(s) is/are required and beneficial for the diagnosis and treatment of an individual’s illness.
- For the diagnosis or prognosis of individuals with confirmed acute leukemia, global/gene specific methylation, microRNA (miRNA) expression, or gene expression analysis is considered NOT MEDICALLY NECESSARY.
- For diagnosis or prognosis of myeloid or lymphoid neoplasms, all other testing not addressed above is considered NOT MEDICALLY NECESSARY.
NOTES:
Note: For two or more gene tests being run on the same platform, please refer to CAM 235- Reimbursement Policy.
Table of Terminology
| Term |
Definition |
| ABL1 |
Abelson murine leukemia viral oncogene homolog 1 |
| ACAs |
Additional chromosomal abnormalities |
| ALL |
Acute lymphoblastic leukemia |
| AML |
Acute myeloid leukemia |
| ANKRD26 |
Ankyrin repeat domain containing 26 |
| AP |
Accelerated phase |
| AP-CML |
Accelerated phase-chronic myeloid leukemia |
| APL |
Acute promyelocytic leukemia |
| ASCO |
American Society of Clinical Oncology |
| ASH |
American Society of Hematology |
| ASXL1 |
ASXL transcriptional regulator 1 |
| ASXL2 |
ASXL transcriptional regulator 2 |
| BAALC |
Brain and acute leukemia cytoplasmic |
| B-ALL |
B-cell acute lymphoblastic leukemia |
| BCR-ABL1 |
BCR activator of RhoGEF and GTPase- ABL proto-oncogene 1, non-receptor tyrosine kinase |
| BCSH |
British Committee for Standards in Haematology |
| BLNK |
B-cell linker |
| BM |
Bone marrow |
| BP |
Blast phase |
| BPDCN |
Blastic plasmacytoid dendritic cell neoplasm |
| c-KIT |
Commonly used alias for KIT proto-oncogene, receptor tyrosine kinase |
| CAP |
College of American Pathologists |
| CBC |
Complete blood count |
| CBF |
Core-binding factor |
| CALR |
Calreticulin |
| CBFA2 |
CBFA2/RUNX1 partner transcriptional co-repressor 2 |
| CCyR |
Complete cytogenetic response |
| CEBPA |
CCAAT/enhancer binding protein alpha |
| cGH |
Comparative genomic hybridization |
| CK |
Complex karyotype |
| CLIA ’88 |
Clinical Laboratory Improvement Amendments of 1988 |
| CLL |
Chronic lymphocytic leukemia |
| CMA |
Chromosomal microarray |
| CMS |
Centers for Medicare and Medicaid |
| CML |
Chronic myeloid leukemia |
| CMP |
Comprehensive metabolic panel |
| CN |
Normal karyotypes |
| CP-CML |
Chronic phase-chronic myeloid leukemia |
| CR |
Complete hematologic remission |
| CT |
Computed tomography |
| CRLF2 |
Cytokine receptor-like factor 2 |
| CSF1R |
Colony stimulating factor 1 receptor |
| CV |
Coefficient of variation |
| CXCR4 |
C-X-C chemokine receptor type 4 |
| DDX41 |
DEAD-box helicase 41 |
| d17p |
Deletion of chromosome 17p |
| DNA |
Deoxyribonucleic acid |
| DNMT3A |
Deoxyribonucleic acid methyltransferase 3A |
| dPCR |
Digital polymerase chain reaction |
| EBF1 |
Early B cell factor-1 |
| ELN |
European Leukemia Net |
| EMSO |
European Society For Medical Oncology |
| EPOR |
Erythropoietin receptor |
| ERG |
ETS transcription factor ERG |
| EVI1 |
Ecotropic virus integration site 1 protein homolog |
| EZH2 |
Enhancer of zeste 2 polycomb repressive complex 2 subunit |
| FCI |
Flow cytometric immunophenotyping |
| FDA |
Food and Drug Administration |
| FDG-PET |
Fluorodeoxyglucose positron emission tomography |
| FISH |
Fluorescence in situ hybridization |
| FLT3 |
FMS-like tyrosine kinase 3 |
| FNA |
First-needle aspiration |
| GATA2 |
GATA binding protein 2 |
| HCT |
Hematopoietic cell transplantation |
| IDH1 |
Isocitrate dehydrogenase 1 |
| IDH2 |
Isocitrate dehydrogenase 2 |
| IGHV |
Immunoglobulin heavy chain variable region gene |
| IHC |
Immunohistochemistry |
| IKZF1 |
Ikaros zinc finger 1 |
| IL7R |
Interleukin-7 receptor |
| ITD |
Internal tandem duplications |
| iwCLL |
International Workshop on Chronic Lymphocytic Leukemia |
| JAK2 |
Janus kinase 2 |
| JNK |
c-Jun N-terminal kinase |
| KIT |
KIT proto-oncogene, receptor tyrosine kinase |
| KRAS |
KRAS proto-oncogene, GTPase |
| LAIP |
Leukemia-associated immunophenotypes |
| LCDs |
Local Coverage Determinations |
| LDH |
Lactate dehydrogenase |
| LOD |
Limit of detection |
| LTD |
Laboratory-developed test |
| MBL |
Monoclonal B lymphocytosis |
| MBNL1 |
Muscleblind like splicing regulator 1 |
| MCL |
Mantle cell lymphoma |
| MDNs |
Myelodysplastic neoplasms |
| MECOM |
MDS1 and EVI1 complex locus |
| MEIS1 |
Muscleblind like splicing regulator 1 |
| MFC |
Multiparameter flow cytometry |
| miRNA |
Micro ribonucleic acid |
| Mkneg |
Non-chromosomal karyotype |
| MLL-PTD |
Mll -Partial Tandem Duplication |
| MMR |
Major molecular response |
| MPL |
MPL proto-oncogene, thrombopoietin receptor |
| MRD |
Minimal residual disease |
| MYD88 |
Myeloid differentiation primary response 88 |
| MYH11 |
Myosin heavy chain 11 |
| EMZL |
Extranodal marginal zone lymphoma |
| NCCN |
National Comprehensive Cancer Network |
| NF1 |
Neurofibromin 1 |
| NGS |
Next generation sequencing |
| NOTCH1 |
Neurogenic locus notch homolog protein 1 |
| NPM1 |
Nucleophosmin 1 |
| NPM1mut |
NPM1 mutations |
| NRAS |
NRAS proto-oncogene, GTPase |
| NTRK3 |
Neurotrophic tyrosine receptor kinase 3 |
| OS |
Overall survival |
| PB |
Peripheral blood |
| PCR |
Polymerase chain reaction |
| PDGFRB |
Platelet-derived growth factor receptor beta |
| PEDALL |
Pediatric Acute Lymphoblastic Leukemia |
| Ph |
Philadelphia |
| PI3K |
Phosphatidylinositol 3-kinase |
| PMF |
Polyamine modulated factor |
| PML-RAR |
Promyelocytic leukemia-retinoic acid receptor |
| PT |
Prothrombin time |
| PTT |
Partial thromboplastin time |
| PTK2B |
Protein tyrosine kinase 2 beta |
| PTPN11 |
Protein phosphatase non-receptor type 11 |
| RNA |
Ribonucleic acid |
| RT-PCR |
Reverse transcription- polymerase chain reaction |
| RUNX1 |
RUNX family transcription factor 1 |
| RUNX1T1 |
RUNX1 partner transcriptional co-repressor 1 |
| sDMR |
Sustained deep molecular response |
| SF3B1 |
Splicing factor 3b subunit 1 |
| SH2B3 |
SH2B adapter protein 3 |
| SH3 |
Src homology 3 |
| SPI1 |
Spi-1 proto-oncogene |
| SRSF2 |
Serine and arginine rich splicing factor 2 |
| SLL |
Small lymphocytic leukemia |
| STAT5 |
Signal transducer and activator of transcription 5 |
| TET2 |
Tet methylcytosine dioxygenase 2 |
| TFR |
Treatment-free remission |
| TKD |
Tyrosine kinase domain |
| TKI |
Tyrosine Kinase Inhibitor |
| T-LGLL |
T-cell large granular lymphocyte leukemia |
| TP53 |
Tumor protein 53 |
| TYK2 |
Tyrosine kinase 2 |
| VAF |
Variant allele frequency |
| WGS |
Whole-genome sequencing |
| WHO |
World Health Organization |
| WT1 |
Wilms' tumor 1 |
Rationale
Acute Lymphoblastic Leukemia (ALL)
Acute lymphocytic leukemia occurs when a bone marrow cell develops a mutation in its genetic material. ALL can be of B-cell precursor (BCP) lineage (BCP-ALL) or, less commonly, T-cell precursor lineage (T-ALL). However, “Both comprise multiple subtypes commonly defined by structural chromosomal alterations that are initiating lesions, with secondary somatic (tumor-acquired) DNA copy number alterations and sequence mutations that contribute to leukemogenesis. Chromosomal alterations include aneuploidy and chromosomal rearrangements that result in oncogene deregulation or expression of chimeric fusion genes. The prevalence of these alterations varies according to age and identification is important for diagnosis, risk classification, and, for some lesions, targeted therapy. Common genomic features of BCR-ABL1–like ALL are alterations of B-lymphoid transcription factor genes (particularly IKZF1 deletions) and genetic alterations deregulating cytokine receptor and tyrosine kinase signaling. These include rearrangements and mutation of CRLF2 (approximately 50%), rearrangements of ABL-class tyrosine kinase genes (12%), rearrangements of JAK2 (7%) and the erythropoietin receptor gene (EPOR; three to ten percent), mutations activating JAK-STAT signaling (11%) and Ras signaling (NRAS, KRAS, PTPN11, and NF1; six percent), and less common kinase alterations (FLT3, NTRK3, BLNK, TYK2, and PTK2B).”4
Acute myeloid leukemia (AML)
Acute myeloid leukemia is the most common acute leukemia in adults (80%), with a median age at diagnosis of 65 years. An AML diagnosis in adults is generally associated with a poor prognosis. This disease is much less common in children younger than ten years of age, as less than ten percent of acute leukemias are diagnosed as AML in this age group.5-7
A clinical presentation of AML includes symptoms related to complications of pancytopenia, including weakness and fatigability, infections of variable severity, and hemorrhagic findings.7 Analysis of gene sequencing of AML cases generally reveal more than ten significant gene mutations; many of which are thought to participate in leukemogenesis.8 The most common gene mutations are as follows: FLT3 (28%), NPM1 (27%), DNMT3A (26%), IDH1 or IDH2 (20%), NRAS or KRAS (12%), RUNX1 (10%), TET2 (8%), TP53 (8%), CEBPA (6%), and WT1 (6%). Mutations impacting signal activation are frequent (60% of cases); the most common of which are mutations in FLT3.9
The second most common mutation in AML (27%) is of nucleophosmin (NPM1), a ubiquitously expressed phosphoprotein that normally shuttles between the nucleus and cytoplasm.9,10 NPM1 mutations are associated with improved outcomes although the mechanism is not known. Concurrent mutations (such as an FTL3 mutation) may influence prognosis, but generally NPM1 patients without concurrent mutations have better prognoses.11
The CCAAT/enhancer binding protein alpha (CEBPA) gene mutation is also common in AML. CEBPA encodes a transcription factor necessary for myeloid differentiation. This mutation is one of the two mutations associated with familial leukemia and consists of about ten percent of AML cases. Familial AML with a CEBPA mutation has a phenotype similar to those of “sporadic AML with biallelic CEBPA mutations,” but most of the current data revolves around the assessment of CEBPA double mutations. CEBPA single mutations and hypermethylated CEBPA requires further study.11
Isocitrate dehydrogenase (IDH) 1 and 2 mutations comprise approximately 15% of AML cases. These mutations are mutually exclusive with Tet Methylcytosine Dioxygenase 2 (TET2) and Wilms' tumor 1 (WT1) mutations but are commonly seen with NPM1 and DNA methyltransferase 3A (DNMT3A) mutations. Data on the prognoses of these mutations is varied.11
The KIT mutations also comprise about six percent of AML cases. KIT encodes the receptor for a stem cell factor, and prognoses are varied.11,12 Some researchers suggest that of all KIT mutations, the D816 mutation is the most unfavorable prognostic factor in AML patients.13
Approximately eight percent of AML cases consist of WT1 gene mutations. WT1 encodes a transcriptional regulator for genes involved in maturation and growth. Again, the prognosis of this mutation is mixed,11 although some researchers strongly support the theory that WT1 mutations are associated with poor AML prognoses.14 The WT1 mutation status of AML patients may also change during disease progression. New research has suggested that after allogenic stem cell transplantation, AML relapse could be due to a gain in WT1 gene alterations and a “high mutation load.”15
The ASXL Transcriptional Regulator 1 (ASXL1) and ASXL Transcriptional Regulator 2 (ASXL2) may also be mutated in AML cases. ASXL1 has an unclear function, but it is speculated to be related to histone post-translational modifications. The frequency of ASXL1 is varied, as estimates range from six percent to 30%. Furthermore, ASXL1 mutations are mutually exclusive with NPM1 mutations, and ASXL2 mutations are associated with RUNX1 mutations (also known as AML1 or CBFA2).11
The DNMT3A gene amounts to 20%-22% of AML cases. This gene plays a role in epigenetic modifications for development and differentiation. Mutations in this gene affect hematopoietic stem cell differentiation. Prognoses of this gene mutation have been mixed.11
Tumor protein 53 (TP53) and RAS and may also be present in AML cases and may be accompanied by other genetic abnormalities. RAS regulates cell signal transduction, and its mutation leads to a constitutionally active growth stimulus whereas TP53 encodes a transcriptional activator of growth inhibitory genes.16,17
Gene expression profiling and microRNA expression profiling may also contribute to assessment and management of AML. Gene expression profiling has been used to differentiate between risk groups based on cytogenetic evaluation whereas microRNA profiling evaluates the regulation of gene expression. However, neither technique is used regularly in clinical practice as these techniques have yet to be widely validated.11
Chronic Lymphoblastic Leukemia (CLL)
In Western countries, CLL is the most common form of leukemia; in the United States, it accounts for 25 to 30 percent of all leukemias.18 CLL and SLL (small lymphocytic leukemia) are mature B cell neoplasms characterized by a progressive accumulation of monoclonal B lymphocytes. It should be noted that “CLL is considered to be identical (ie, one disease with different manifestations) to the non-Hodgkin lymphoma SLL. The malignant cells seen in CLL and SLL have identical pathologic and immunophenotypic features. The term CLL is used when the disease manifests primarily in the blood, whereas the term SLL is used when involvement is primarily nodal.”18 It is also worth mentioning that “In less than 10 percent of patients, CLL/SLL presents primarily in the lymph nodes as a non-Hodgkin lymphoma (ie, SLL)” and that “this presentation accounts for less than 5 percent of all non-Hodgkin lymphomas.”18
The CLL exomes carry approximately “5–20 somatically acquired mutated genes per individual case, much fewer than many other solid cancers. Importantly, the somatically acquired mutation status of sequence variants in genes, like other studies of genomic changes in cancer, necessitates confirmation of the absence of these candidate mutations in paired non-tumorous DNA (T-cell–, buccal cell– or skin-derived DNA). Mutations in TP53 are of major clinical relevance, are often associated with del17p and gain in frequency over time. TP53 mutated and associated del17p states substantially lower response rates, remission duration, and survival in CLL. Mutations in NOTCH1 and SF3B1 are recurrent, often associated with progressive CLL.”19
Chronic myeloid leukemia (CML)
The first human malignancy in which a specific chromosomal defect, known as the minute or Philadelphia (Ph) chromosome, could be linked to pathogenetic events of leukemogenesis.20 The Ph chromosome translocation (t(9;22)(q34;q11.2)) fuses the breakpoint cluster region protein (BCR) gene from chromosome 22 with the Abelson murine leukemia viral oncogene homolog 1 (ABL1) proto-oncogene from chromosome nine in a head-to-tail manner to form the transcriptionally active BCR-ABL fusion gene.21 The fusion of BCR at the 5’ side in ABL alters the tightly regulated function of the Src homology 3 (SH3) domain, disabling control over the tyrosine kinase enzyme. The resulting chimeric BCR-ABL protein has constitutively elevated tyrosine phosphokinase activity22 that activates a number of downstream signaling molecules, including PI3K, AKT, JNK, RAS and STAT5.23 This then disrupts cellular signal transduction pathways, leading to issues in the regulation of both apoptosis and cell proliferation,24 ultimately leading to factor-independent and leukemogenic cell growth.25
Detection of the Ph chromosome is the hallmark of CML and is found in up to 95 percent of patients.26 In approximately five percent of CML cases, the Ph chromosome cannot be detected, and BCR-ABL1 formation is attributed to microscopically undetectable translocations or variant complex translocations involving a third chromosome.21 Independent of which other chromosomes are involved in variant translocations, the generation of the BCR-ABL fusion gene is the “fundamental cause of Ph-positive leukemias,”25 as the 210-KDa fusion protein BCR-ABL is essential for initiation, maintenance, and progression of CML.23 Testing for BCR-ABL1 detects both the Ph chromosome and fusion gene or its transcripts. The BCR-ABL1 transcript is the RNA copy made by the cell.
The FMS-like tyrosine kinase 3 (FLT3) is a transmembrane tyrosine kinase receptor that stimulates cell proliferation upon activation. Both internal tandem duplications (ITDs) of different lengths and point mutations in the activating loop of the kinase domain result in ligand-independent activation of the FLT3 receptor and a proliferative signal. A FLT3-ITD mutation has been shown to have a poor prognosis in contrast to FTL3 point mutations in the activation loop of the kinase domain. Higher ratios of mutated alleles compared to wild-type alleles confer worse prognoses.11
The BCR-ABL1, refers to the fusion gene resulting from a reciprocal translocation that joins the ABL1 gene from chromosome 9 to the BCR gene on chromosome 22 and is necessary for the development of chronic myeloid leukemia.25,27 This reciprocal translocation also generates a shortened derivative chromosome 22, known as the Philadelphia (Ph) chromosome.21 The Ph chromosome is a diagnostic hallmark, present in 95% of people with CML and approximately three to five percent children and 25%–40% adults with acute lymphoblastic leukemia (ALL).26 An aggressive form of cancer resulting from the neoplastic transformation of lymphoid precursors characterized by the presence of too many lymphoblasts or lymphocytes in the bone marrow and peripheral blood.28 Predominately a childhood disease, approximately 60% of cases were diagnosed in patients younger than 20 years of age.29
Analytic Validity
There is very limited published literature on the analytic validity and clinical validity of genetic testing for FLT3 and NPM1 mutations in AML. However, the analytic validity of PCR in general is extremely high.30 Other tools, such as flow cytometry and next generation sequencing (NGS) have also been used for AML prognostic and diagnostic purposes.
Ampasavate, et al. (2019) have developed a quantitative protocol and flow cytometry-based method for monitoring an anti-FLT3 interaction. The FLT3 biomarker has been previously identified as a poor prognostic marker for AML patients. This method can rapidly identify intact FLT3 on the leukemic cell surface. “The results demonstrated good linearity (r2 > 0.99)”; further, “when compared with Western blotting results, FLT3 protein expression levels in leukemia patient's bone marrow samples were demonstrated in the same trend.”31 The researchers state that this technique is reliable, rapid, effective and “provided a practical analysis of FLT-3 biomarker levels which is valuable for physician decision in acute leukemia treatment.”31
Alonso, et al. (2019) have researched the utility of a 19-gene NGS panel for AML diagnostic purposes. This targeted NGS panel was studied in a cohort of 162 patients with AML. The authors note that “the assay yielded a median read depth >2000×, with 88% of on-target reads and a mean uniformity >93% without significant global strand bias. The method was sensitive and specific, with a valid performance at the clinical variant allele frequency cutoff of 3% for point mutations and five percent for insertions or deletions.” The researchers conclude that this is a “reliable and reproducible method” for AML diagnoses.32
Molecular testing for the diagnosis of CML confirms typical findings in the blood and bone marrow by the demonstration of the Ph chromosome, the BCR-ABL1 fusion gene or the BCR-ABL1 fusion mRNA. Molecular testing techniques include conventional cytogenetics, fluorescence in situ hybridization (FISH) analysis and reverse transcription polymerase chain reaction (RT-PCR).25 Conventional cytogenetic karyotyping is no longer the diagnostic modality of choice due to its requirements for a highly skilled staff, culturing of cells, long turnaround time, and lower sensitivity (5-10%). Despite this, conventional cytogenetics are still the gold standard, and “should be performed” especially at diagnosis to detect additional clonal abnormalities.33 FISH is more sensitive (0.1-5%) than karyotyping and can be performed on peripheral blood in addition to bone marrow and tissue. FISH can detect certain very rare translocations not usually detectable by the vast majority of commercial and laboratory-developed RT-PCR assays, but FISH is highly specific to the targeted region and may miss other chromosomal changes. Quantitative RT-PCR is the most sensitive technique currently available (0.001-0.01% sensitivity).
On July 22, 2016 the FDA approved the QuantideX qPCR BCR-ABL IS Kit as an in vitro nucleic acid amplification test for the quantitation of BCR-ABL1 and ABL1 transcripts in total RNA from whole blood of diagnosed t(9;22) positive Chronic Myeloid Leukemia (CML) patients expressing BCR-ABL1 fusion transcripts type e13a2 and/or e14a2 as a class II device with special controls.34 Brown, et al. (2019) performed a study to describe the analytical validation of this kit. They were able to find that “the test has a limit of detection of MR molecular response] 4.7 (0.002% IS) and a linear range from MR0.3 (50%IS) to MR4.7 (0.002%IS) for both Major transcripts. Single-site and multisite precision studies demonstrated a maximum SD of 0.13 MR (30% CV within the assay range between MR0.7 and MR3.7).”
Clinical Utility and Validity
The clinical utility of testing fallows for further risk stratification, prognostication, and guide management decisions in patients with AML. Several studies have concluded that FLT3 and NPM1 mutation testing in cytogenetically normal AML is useful for prognosis and treatment decision making.36-38
Devillier, et al. (2015) sought to identify biological and prognostic subgroups based on genetic mutations in AML patients. A total of 125 AML patients with myelodysplasia-related changes (“MRC”) were evaluated. The authors focused on the 26 patients with ASXL1 mutations and 28 with TP53 mutations. The ASXL1 mutation cohort was found to have a higher proportion of marrow dysgranulopoiesis and an overall survival (OS) rate that was below average for wild-types (14% for ASXL1 mutants, 37% for wild-types). The TP53 cohort was found to have a “complex karyotype” and predicted a poor outcome with unfavorable cytogenetic risk AML. Both mutations were found to be an independent factor associated with shorter OS.39
Bolouri, et al. (2018) examined 993 children’s genetic data from the Children's Oncology Group (COG) AML trials to characterize the molecular landscape of AML. The authors found that certain somatic variants, such as MBNL1, were “disproportionately prevalent” in children compared to adults. However, certain variants common in adults such as TP53, were not found in children. Other mutations such as NRAS and KRAS were “frequent” in pediatric AML. The authors concluded that their results “highlight the need for and facilitate the development of age-tailored targeted therapies for the treatment of pediatric AML.”40
Jongen-Lavrencic, et al. (2018) conducted a study of 482 patients 18 to 65 years with newly diagnosed AML. Targeted next-generation sequencing (NGS) was carried out at diagnosis and after induction therapy (during complete remission). At least one mutation was detected in 430 (89.2%) patients, and mutations persisted in 51.4% of those patients during complete remission. The detection of minimal residual disease was associated with a significantly increased relapse rate than no detection. Persistent DTA mutations (mutations in DNMT3A, TET2, and ASXL1) were not correlated with an increased relapse rate. Overall, the authors concluded, “a comparison of sequencing with flow cytometry for the detection of residual disease showed that sequencing had significant additive prognostic value. Among patients with AML, the detection of molecular minimal residual disease during complete remission had significant independent prognostic value with respect to relapse and survival rates, but the detection of persistent mutations that are associated with clonal hematopoiesis did not have such prognostic value within a 4-year time frame.”41
Kuwatsuka, et al. (2018) evaluated the genetic background of 103 young adults and their subsequent clinical outcomes. The 103 cases included mutations in FLT3-ITD, KIT, CEBPA, NRAS, KRAS, WT1, MLL-PTD, and NPM1. Overall, FLT3-ITD and NPM1 mutations were associated with a greater mortality risk. NPM1 mutations conferred a 100% survival rate in the absence of FLT3-ITD mutations, but FLT3-ITD conferred only a 35% survival without NPM1 mutations.42
Zhu, et al. (2017) assessed the effect of gene mutations on the subsequent cytogenetic aberrations. A total of 560 patients were enrolled, and the authors examined the following alterations: “CEBPA, NPM1, FLT3, C-KIT, NRAS, WT1, DNMT3A, MLL-PTD and IDH1/2, as well as expression levels of MECOM, ERG, GATA2, WT1, BAALC, MEIS1 and SPI1.” The investigators found that the expression levels of MECOM, MEIS1, and BAALC influenced cytogenetic aberration. Further, FLT3, C-KIT, and NRAS mutations all contained a “conversed” expression profile of MEIS1, WT1, GATA2, and BAALC expression. The investigators also noted “FLT3, DNMT3A, NPM1 and biallelic CEBPA represented the mutations associated with the prognosis of AML in our group.”43
Papaemmanuil, et al. (2016) examined the relationship between genotype and pathophysiology in AML. A total of 1540 patients with 5234 driver mutations across 76 genes were studied. The authors found three genomic subcategories in addition to the currently defined AML subgroups: mutations in genes encoding chromatin, RNA splicing regulators or both (such as ASXL1 or RUNX1), TP53 mutations, chromosomal aneuploidies or both (unusual karyotypes and TP53), and IDH2 mutations. The authors noted that “patients with chromatin–spliceosome and TP53–aneuploidy AML had poor outcomes.” The NPM1 cohort was the largest of the sample (27%, 436 patients) and 319 of those patients also carried a DNA methylation or hydroxylmethylation gene, such as IDH1/2 or TET2. The authors also noted that NPM1-DNMT3A–NRASG12/13 had an “unexpectedly benign” prognosis, and the NPM1 subgroup’s prognoses were largely determined by the context in which the NPM1 mutation occurred (such as in NRAS, IDH, and so on).44
Sperr, et al. (2016) evaluated the effect of a genetic mutation and karyotype on the efficacy of treatment for elderly patients. A total of 192 patients over 60 years old were enrolled, and 115 of these patients achieved “complete hematologic remission (CR).” The authors stated that NPM1 mutations (NPM1mut) and karyotype were the only independent predictors of survival, also noting that NPM1mut showed a prognostic impact on both normal (CN) and non-chromosomal (Mkneg) karyotypes. The authors concluded that “elderly patients with CN/Mkneg-NPM1mut or core binding factor AML can achieve long term median continuous CR when treated with intensive induction and consolidation therapy whereas most elderly patients with CN/Mkneg-NPM1wild-type or CN/Mkpos AML may not benefit from intensive chemotherapy.”45
Heiblig, et al. (2019) assessed the impact of NPM1 subtypes on treatment outcomes. One hundred seventy-five patients were examined. The authors found that out of the NPM1 AML cases, 73% (128) were “Type A” mutations (TCTG at exon 12) and 27% (47) were “non Type-A mutations” (Type B: CATG and Type D: CGTG). The Type-A mutations were found to achieve minimal residual disease (MRD) earlier than non Type-A mutations. However, non-type A mutations achieved better rates of medial survival.46
D’Adda, et al. (2019) evaluated the effect of the BCR-ABL transcript on efficacy of TKIs. The BCR-ABL1 fusion gene may cause CML pathogenesis due to several breakpoints; the most common occur around exon 13 and 14 of the BCR gene and cause the formation of e13a2 and e14a2 transcripts.48 Out of 173 sampled patients, 67 had the e13a2 transcript, and 106 had the e14a2 version. The patients with the e14a2 version were more likely to achieve a deep molecular response to TKIs (sustained or otherwise). After 68 months, the sustained deep molecular response (sDMR) rate was 39.6% for e14a2 patients compared to 19.6% for e13a2 patients. Overall, the maximum rate of sDMR for e13a2 patients was 37%, after 60 months. Furthermore, only two patients (three percent) with the e13a2 transcript achieved treatment-free remission (TFR) whereas 25 of e14a2 patients achieved TFR (23%).47
Xu, et al. (2020) have analyzed data from 220 normal karyotype AML pediatric patients. Participants were selected from the Cancer Genome Atlas database. It was found that 12.7% of these patients had WT1 mutations, and that “the WT1-mutated group suffered lower rates of complete remission (CR) (P < 0.001 and P < 0.001, respectively) but higher rates of minimal residual disease (MRD) (P = 0.003 and P = 0.021, respectively) after both one and two courses of induction chemotherapy.”49 Patients with WT1 mutations also had significantly worse event-free and overall survival rates (P=0.007 and P<0.001, respectively).49
Sasaki, et al. (2020) studied the impact of ASXL1, DNMT3A, JAK2, TET2, and TP53 mutations on survival in newly diagnosed acute myeloid leukemia (AML) patients. The 421 bone marrow aspirates were studied using NGS for these mutations with a minimum variant allele frequency (VAF) of five percent. "A total of 71 patients (17%) had ASXL1 mutations, 104 patients (25%) had DNMT3A mutations, 16 patients (4%) had JAK2 mutations, 82 patients (20%) had TET2 mutations, and 86 patients (20%) had TP53 mutations." The median VAF of ASXL1 was 34.31% (range, 1.17%‐58.62%), DNMT3A was 41.76% (range, 1.02%‐91.66%), JAK2 was 46.70% (range, 10.4%‐71.7%), TET2 was 42.78% (range, 2.26%‐95.32%), and TP53 was 45.47% (range, 1.15%‐93.74%). In patients with these mutations, the median overall survival was 11 months. In patients without these mutations, the overall survival was 27 months. The authors conclude that "The VAF of ASXL1, DNMT3A, JAK2, TET2, TP53, and NPM1 mutations is associated with worse prognosis in patients with newly diagnosed AML."50
Duncavage, et al. (2021) investigated the clinical utility and accuracy of whole-genome sequencing (WGS) for the purpose of risk stratification in patients with acute myeloid leukemia (AML) or myelodysplastic syndromes (MDS). The results from 263 patients were compared with findings from cytogenetic analysis and targeted sequencing. When conducting the WGS, they found that all 40 recurrent translocations and 91 copy-number alterations found on cytogenetic analysis were identified on WGS. There were also new clinically reportable genomic events among 17.0% of the patient sample. The standard AML risk groups, “as defined by sequencing results instead of cytogenetic analysis, correlated with clinical outcomes.” WGS was also able to classify patients with inconclusive cytogenetic analysis results into risk groups. This demonstrates that WGS could increase the diagnostic yield of AML and MDS and supplement the accuracy of cytogenetic analysis.51
In a retrospective study, Yang reported the clinical significance of BCR-ABL1 mutations in patients with Philadelphia chromosome-positive CML who underwent allogeneic hematopoietic cell transplantation. Of 315 patients in this study, point mutations were detected in 152 patients. One hundred and one of these 152 patients (66%) had T315I mutations and 51 had mutations other than T315I. The patients were followed 38 months later and overall survival at three years was worse in patients with the mutations than the non-mutation group, especially in the chronic phase of CML. Overall survival in the non-T315I group was significantly worse than that in the no-mutation group. The authors conclude that "mortality risk was significantly higher in patients with the BCR-ABL1 mutation than in patients without the mutation.”27
World Health Organization (WHO)
In a revision of the fourth edition on Classification of Tumors of Hematopoietic and Lymphoid Tissues, World Health Organization (WHO) incorporated new molecular genetic findings and clinical data into its classification of acute leukemias. WHO expanded on the prognostic significance of various gene mutations for each AML subtype. For example, for the AML with recurrent genetic abnormalities, inv(3) (q21.3; q26.2) does not represent a fusion gene, but rather a repositioning of a distal GATA2 enhancer leading to activation of MECOM (EVI1) expression and GATA2 haploinsufficiency. The AML with CEBPA mutation is defined based on biallelic mutation instead of single mutations because of prognostic significance. The provisional two categories are also added such as AML with RUNX1 for de novo AML without preexisting cytogenetic abnormalities associated with MDS and AML with BCR-ABL1 fusion gene. AML with NPM1 or biallelic CEBPA mutations and multilineage dysplasia are now considered separately instead of being a part of AML with myelodysplasia-related changes because of a lack of prognostic significance. The complete list for acute myeloid neoplasms 2016 WHO classification is shown in Figure 1.52
In the absence of JAK2, CALR, and MPL mutations, the presence of another clonal marker is included as one of the major diagnostic criteria for PMF. Additional mutation in ASXL1, EZH2, TET2, IDH1, IDH2, SRSF2 and SF3B1 genes are noted to be of use in determining the clonal nature of the disease.
In the fifth edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms, WHO places greater emphasis in “the separation of AML with defining genetic abnormalities from AML defined by differentiation.” The latter eschews the previously ambiguous categorical term AML NOS, and other notable changes include the “elimination of the 20% blast requirement for AML types with defining genetic abnormalities (with the exception of AML with BCR::ABL1 fusion and AML with CEBPA mutation)” and “the introduction of a section on AML with other defined genetic alterations, a landing spot for new and/or uncommon AML subtypes that may (or may not) become defined types in future editions of the classification.”53 Their list of acute myeloid leukemias is presented below.53


Moreover, in defining acute myeloid leukemia, myelodysplasia-related (AML-MR), WHO offers the following abnormalities and mutations of interest:53


The WHO published guidelines on the classification of myeloid neoplasms and acute leukemia. In the recent revision of the fourth edition on Classification of Tumors of Hematopoietic and Lymphoid issues, World Health Organization (WHO) incorporated new molecular genetic findings and clinical data into its classification of acute leukemias.52
They state that: “With regard to chronic myeloid leukemia , BCR-ABL1+, most cases of CML in chronic phase can be diagnosed from peripheral blood (PB) findings combined with detection of t(9;22)(q34.1;q11.2) or, more specifically, BCR-ABL1 by molecular genetic techniques. However, a bone marrow (BM) aspirate is essential to ensure sufficient material for a complete karyotype and for morphologic evaluation to confirm the phase of disease. In the era of tyrosine-kinase inhibitor (TKI) therapy, newly diagnosed patients may have a nearly normal lifespan, but regular monitoring for BCR-ABL1 burden and for evidence of genetic evolution and development of resistance to TKI therapy is essential to detect disease progression.”52
They also introduced a provisional classification of ALL: B-ALL with translocations involving tyrosine kinases or cytokine receptors (“BCR-ABL1–like ALL”). They note that “this newly recognized entity is assuming increasing importance because of its association with an adverse prognosis and responses of some cases to TKI therapies; however, it has been difficult to define in the clinical setting. It was originally described separately by different groups who demonstrated a series of cases of poor-prognosis childhood ALL with gene expression profiles similar to those seen in cases of ALL with BCR-ABL1, though different algorithms applied to the same sets of cases did not classify all cases the same way.
The cases with translocations involving tyrosine kinase genes involve many different genes including ABL1 (with partners other than BCR), as well as other kinases including ABL2, PDGFRB, NTRK3, TYK2, CSF1R, and JAK2. Over 30 different partner genes have been described. Some patients, especially those with EBF1-PDGFRB translocations, have shown remarkable responses to TKI therapy, even after failing conventional therapy.”52
In the 5th Edition of the World Health Organization Classification of Hematolymphoid Tumors, the WHO updated their cytogenetic and molecular genetic information regarding AML. They highlight the following in their update:
- “Newly discovered recurrent cytogenetic abnormalities listed in AML with other defined genetic abnormalities include RUNX1T3 (CBFA2T3)::GLIS, KAT6A::CREBBP, FUS::ERG, MNX1::ETV6, and NPM1::MLF1. Those listed in “B-LBL/L with other defined genetic abnormalities” include DUX4 rearrangement, MEF2D rearrangement, ZNF384 rearrangement, NUTM1 rearrangements, MYC rearrangement, PAX5alt or PAX5 p.P80R. With more data accumulated, neoplasms with these new genetic abnormalities will very likely become separated subtypes in the next edition, and there will be more newly discovered genetic alterations listed here. Biallelic TP53 alterations have been frequently found in MDNs, acute erythroid leukemia, and myeloid neoplasms post cytotoxic therapy, and they usually predict a worse prognosis”
- “Gene mutations in the MAPK pathway are commonly seen in histiocytic neoplasms. CXCR4 mutations are detected in a significant proportion of lymphoplasmacytic lymphoma cases, mostly concurrent with MYD88 mutations, and the presence of them is associated with resistance to ibrutinib therapy (32). The mutational profiles of extranodal MZL (EMZL) and nodal MZL differ, and there are significant genetic differences among EMZLs arising in different anatomic sites”
- “STAT3 mutation is commonly seen in CD8+ T-LGLL and gamma/delta T-LGLL, and it is associated with neutropenia and unfavorable overall survival; while STAT5b mutation is associated with a poor prognosis only in CD8+ T-LGLL but has no prognostic impact in CD4+ T-LGLL and gamma/delta T-LGLL.”54
Though the WHO did not take any specific stance with these in their new version, these mutations and pathways may be worth keeping abreast of for the future.
College of American Pathologists (CAP) and American Society of Hematology (ASH)
Following recent progress in molecular genetic findings and 2016 WHO classification of acute leukemias, the College of American Pathologists (CAP) and the American Society of Hematology (ASH) have formed an expert panel to review and establish guidelines for appropriate laboratory testing. The published guideline provides twenty-seven guideline statements ranging from recommendations on what testing is appropriate for the diagnostic and prognostic evaluation of leukemias to where the testing should be performed and how results should be reported.
There, the societies recommend that “the pathologist should review recent or concurrent CBC counts and leukocyte differentials and evaluate a peripheral blood (PB) smear (Strong recommendation).” Moreover, they note that “in addition to performing morphologic assessment (blood and BM), the pathologist or treating clinician should obtain sufficient samples and perform conventional cytogenetic analysis (ie, karyotype), appropriate molecular genetic and/or FISH testing, and FCI. The flow cytometry panel should be sufficient to distinguish AML (including APL), including early T-ALL, B-ALL, and AL of ambiguous lineage in all patients diagnosed with AL. Molecular genetic and/or FISH testing does not, however, replace conventional cytogenetic analysis (Strong recommendation).”55
In those patients with suspected or confirmed AL, “the pathologist may request and evaluate cytochemical studies to assist in the diagnosis and classification of AML (Expert consensus opinion).” Furthermore, “for patients with suspected or confirmed AL, the pathologist may use flow cytometry in the evaluation of CSF (Recommendation)” and that “the pathologist or treating clinician should ensure that flow cytometry analysis or molecular characterization is comprehensive enough to allow subsequent detection of MRD (Strong recommendation).” It is also mentioned that “for patients with AL other than those with ALL who are receiving intrathecal therapy, the treating clinician may, under certain circumstances, obtain a CSF sample when there is no clinical contraindication. The treating clinician or pathologist should ensure that a cell count is performed and that examination and enumeration of blasts on a cytocentrifuge preparation are performed and are reviewed by the pathologist (Expert consensus opinion).”55
Many genes should be tested for patients with suspected or confirmed B-ALL. In pediatric patients, “the pathologist or treating clinician should ensure that testing for t(12;21)(p13.2;q22.1); ETV6-RUNX1, t(9;22)(q34.1;q11.2); BCR-ABL1, KMT2A (MLL) translocation, iAMP21, and trisomy 4 and 10 is performed (Strong recommendation),” while for adult patients, they assert “the pathologist or treating clinician should ensure that testing for t(9;22)(q34.1;q11.2); BCR-ABL1 is performed. In addition, testing for KMT2A (MLL) translocations may be performed. (Strong recommendation for testing for t(9;22)(q34.1;q11.2) and BCR-ABL1; Recommendation for testing for KMT2A (MLL) translocations).”55
The CAP and the ASH also propound that “for patients with suspected or confirmed ALL, the pathologist or treating clinician may order appropriate mutational analysis for selected genes that influence diagnosis, prognosis, and/or therapeutic management, which include, but are not limited to, PAX5, JAK1, JAK2, and/or IKZF1 for B-ALL and NOTCH1 and/or FBXW7 for T-ALL. Testing for overexpression of CRLF2 may also be performed for B-ALL (Recommendation).”
The appropriate molecular genetic testing for AML is discussed starting from 16th guideline statement.
The expert panel strongly recommends testing for FLT3-ITD in adult and pediatric patients with suspected or confirmed AML of any type. They also recommend testing for other mutational analysis that could include, but not limited to, IDH1, IDH2, TET2, WT1, DNMT3A, and/or TP53 for prognostic and/or therapeutic purposes (Statement 16).
In the 17th guideline statement, expert panel strongly recommends testing for KIT mutation in adult patients with confirmed core-binding factor (CBF) AML (AML with t(8;21)(q22;q22.1); RUNX1-RUNX1T1 or inv(16)(p13.1q22) /t(16;16)(p13.1;q22); CBFB-MYH11). It is only an expert consensus opinion for testing KIT mutation in pediatric patients with confirmed core binding factor AML (AML with t(8;21)(q22;q22.1); RUNX1- RUNX1T1 or inv(16)(p13.1q22) /t(16;16)(p13.1;q22); CBFB-MYH11) which is not a strong recommendation (Statement 17).
The strong recommendation is also given for patients other than those with confirmed core binding factor AML, APL, or AML with myelodysplasia-related cytogenetic abnormalities that testing is needed for mutational analysis for NPM1, CEBPA, and RUNX1 (Statement 19).
In the 20th guideline statement, expert panel is providing no recommendation on either for or against the use of global/gene specific methylation, microRNA (miRNA) expression, or gene expression analysis for diagnosis or prognosis in patients with confirmed acute leukemia.
Finally, in their last statement, expert panel strongly recommends the use of current WHO terminology for the final diagnosis and classification of acute leukemias.56
American Society of Clinical Oncology (ASCO)
The ASCO fully endorses the 2017 CAP and ASH guideline regarding the initial diagnostic work-up of acute leukemia.55 The ASCO supports all twenty-seven guideline statements. The statements relevant to this policy are noted in the 2017 CAP and ASH guidelines above.
National Comprehensive Cancer Network (NCCN)
Acute myeloid leukemia
For the evaluation of AML in individuals 18 years of age or older, the NCCN guidelines recommend a history and a physical, complete blood count (CBC), platelets, differential, comprehensive metabolic panel (CMP), uric acid, lactate dehydrogenase (LDH), B12 and folic acid evaluation, prothrombin time (PT), partial thromboplastin time (PTT), and fibrinogen as part of the workup. Moreover, they recommend bone marrow core biopsy and aspirate analyses (including immunophenotyping by immunohistochemistry stains plus flow cytometry) and analysis for chromosomal structural variations using cytogenetics, FISH, or whole genome sequencing.
In terms of molecular analyses, they list “ASXL1, BCOR, KIT, EZH2, FLT3 [ITD (internal tandem duplication) and TKD (tyrosine kinase domain)], NPM1, CEBPA, IDH1, IDH2, RUNX1, TP53, and other mutations” as targets. A “comprehensive pathology report, including diagnosis of AML (acute myeloid leukemia) within the context of the diagnostic system used, blast count, cellularity, morphologic dysplasia, and mutation status if available” is also mentioned, and the NCCN adds to “consider additional molecular and genetic testing for heritable hematologic malignancy predisposition in a subset of patients, particularly in patients <50 years and those with a family history.” For patients with potential hematopoietic cell transplantation (HCT) in the future, HLA typing is also noted, “(except for patients with a major contraindication to HCT).”57 The NCCN also highlights that “a variety of gene mutations are associated with specific prognoses (category 2A) and may guide medical decision-making (category 2B). Other genetic lesions may have therapeutic significance. The field of genomics in myeloid malignancies and related implications in AML are evolving rapidly. Mutations should be tested in all patients. Multiplex gene panels and targeted next-generation sequencing (NGS) analysis are recommended for the ongoing management of AML and various phases of treatment.”57
The NCCN cautions that the “importance of obtaining adequate samples of marrow or peripheral blood at diagnosis for full karyotyping and FISH cytogenetic analysis for the most common abnormalities cannot be overemphasized. Although FISH studies for common cytogenetic abnormalities may allow for rapid screening to identify either r favorable-, intermediate-, or poor/adverse-risk groups, additional tests are needed to provide a full picture of the genetic factors that contribute to risk.”57
As a part of monitoring during therapy for individuals with AML, the NCCN recommends a bone marrow aspirate/biopsy 14 to 21 days after the “start of therapy to document hypoplasia. If hypoplasia is not documented or indeterminate, repeat biopsy within 7 days to clarify persistence of leukemia. If hypoplasia, then repeat biopsy at time of hematologic recovery to document remission. If cytogenetics were initially abnormal, include cytogenetics as part of the remission documentation.” Furthermore, as a follow-up to both standard-dose cytarabine induction therapy or high-dose cytarabine induction for poor-risk AML, the NCCN recommends that all patients should have bone marrow aspirate and biopsy by day 42 post-treatment, “regardless of the degree of hematologic recovery. When performed, BM aspirate and biopsy should include cytogenetic and molecular studies, as appropriate.”57
As a part of surveillance and therapy for individuals with relapsed/refractory disease after the completion of consolidation, the NCCN recommends comprehensive genomic profiling to determine mutation status of actionable genes.
For the evaluation or workup for blastic plasmacytoid dendritic cell neoplasm in individuals 18 years of age or older, the NCCN recommends cytogenetic analysis (karyotype and/or FISH) and molecular analysis “(most common aberrations include: TET2, ASXL1, ZRSR2, SRSF2, TP53, NRAS, IDH2, and ETV6).”57
Chronic Lymphocytic Leukemia (CLL) and Small Lymphocytic Leukemia (SLL)
For the diagnosis of chronic lymphocytic leukemia and small lymphocytic leukemia, the NCCN notes the following components as essential:
- “Hematopathology review of peripheral blood smear and all slides with at least one paraffin block representative of the tumor, if the diagnosis was made on a lymph node or bone marrow biopsy.
- Flow cytometry of blood is adequate for the diagnosis of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL).
- CLL diagnosis requires presence of monoclonal B lymphocytes ≥5 x 109/L in peripheral blood based on flow cytometrya
- Clonality of B cells should be confirmed by flow cytometry
- Adequate immunophenotyping to establish diagnosis by flow cytometryb: kappa/lambda, CD19, CD20, CD5, CD23, CD10, CD200; if flow cytometry is used to establish diagnosis, also include cytospin for cyclin D1 or fluorescence in situ hybridization (FISH) for t(11;14); t(11q;v) to exclude mantle cell lymphoma (MCL)
- SLL diagnosis requires presence of lymphadenopathy and/or splenomegaly with monoclonal B lymphocytes ≤5 x 109/L in peripheral blood
- SLL diagnosis should be confirmed by histopathology evaluation of lymph node biopsy
- Biopsy is generally not required. If diagnosis is not established by flow cytometry, then proceed with lymph node biopsy. Bone marrow aspirate with biopsy can be pursued if peripheral blood and lymph node biopsy material are nondiagnostic. Fine-needle aspiration (FNA) or core needle biopsy alone is not generally suitable for the initial diagnosis of CLL/SLL. In certain circumstances, when a lymph node is not easily accessible for excisional or incisional biopsy, a combination of core needle biopsy and FNA biopsy in conjunction with appropriate ancillary techniques for the differential diagnosis (ie, immunohistochemistry [IHC], flow cytometry) may be sufficient for diagnosis.
- Adequate immunophenotyping to establish diagnosis by IHCb: CD3, CD5, CD10, CD20, CD23, cyclin D1, LEF1, SOX11c
- Absolute monoclonal B lymphocyte counta.”58
In superscript a, the NCCN cautions that “absolute monoclonal B lymphocyte count <5000/mm3 that persists more than 3 months in the absence of palpable adenopathy or other clinical features of lymphoproliferative disorder is MBL. Cells of the same phenotype may be seen in reactive lymph nodes; therefore, diagnosis of SLL should only be made when effacement of lymph node architecture is seen. Bone marrow examination is not helpful for the diagnosis of MBL.” Moreover, they note in superscript b that “Typical immunophenotype: CD5+, CD23+, CD43+/-, CD10-, CD19+, CD20 dim, sIg dim+, and cyclin D1-. Note: Some cases may be sIg bright+ or CD23- or dim; some MCL may be CD23+; cyclin D1 immunohistochemistry or FISH for t(11;14) should be considered in all cases, especially for those with an atypical immunophenotype (ie, CD23 dim or negative, CD20 bright, sIg bright). CD200 positivity may distinguish CLL from mantle cell lymphoma (MCL), which is usually CD200.”58
Other analyses that may be “informative for prognostic and/or therapy determination” include
- “FISH to detect: +12; del(11q); del(13q); del(17p)
- TP53 sequencing
- CpG-stimulated metaphase karyotype for complex karyotype (CK)
- Molecular analysis to detect: Immunoglobulin heavy chain variable region gene (IGHV) mutation status
- Beta-2-microglobulin.”58
During workup, history and physical exam including measurement of size of liver and spleen and palpable lymph nodes, performance status, B symptoms, complete blood count (CBC) with differential, comprehensive metabolic panel, and beta-2-microglobulin are essential according to the NCCN. They also note that the following tests may be “useful under certain circumstances”: quantitative immunoglobulins; reticulocyte count, haptoglobin, and direct antiglobulin test (Coombs); chest/abdominal/pelvic CT with contrast of diagnostic quality, if clinically indicated; uric acid; lactate dehydrogenase (LDH); unilateral bone marrow aspirate and biopsy (may be informative for the diagnosis of immune-mediated or disease-related cytopenias); hepatitis B and C testing if treatment is contemplated; pregnancy testing in patients of childbearing age if systemic therapy or RT is planned; discussion of fertility preservation; and FDG-PET/CT scan to direct nodal biopsy, if histologic transformation is suspected.58
Chronic Myeloid Leukemia
The NCCN’s recommendations for CML include the following table:
| Monitoring Response to TKI Therapy and Mutational Analysis |
|
| Test |
Recommendation |
| Hematologic |
CBC every 1–2 weeks for the first 1–2 months (or until stable normalization of blood counts) and thereafter as indicated based on the persistence of cytopenias |
| Bone marrow cytogenetics1 |
At diagnosis Response milestones not reached Any signs of loss of hematologic response Any sign of loss of CCyR or its molecular response correlate (MR2.0: BCR::ABL1 [IS] ≤1%) - defined as an increase in BCR::ABL1 transcript to >1% |
| qPCR using IS [International Scale] |
If there is 1-log increase in BCR::ABL1 transcript levels with MMR, qPCR should be repeated in 1-3 months |
| BCR::ABL kinase domain mutation analysis3 |
Disease progression to AP-CML or BP-CML |
| 1FISH has been inadequately studied for monitoring response to treatment. 2CCyR correlates with BCR-ABL1 (IS) ≤1% (MR2.0). 3Consider myeloid mutation panel to identify BCR::ABL1–independent resistance mutations in patients with no BCR::ABL1 kinase domain mutations |
|
The NCCN (2025) also states that for the diagnosis and workup of CML-1:
- “Initial evaluation should consist of a history and physical exam, including palpation of spleen, complete blood count (CBC) with differential, chemistry profile, and hepatitis B panel. Bone marrow aspirate and biopsy for morphologic and cytogenetic evaluation and quantitative reverse transcriptase polymerase chain reaction (RT-PCR) to establish the presence of quantifiable BCR::ABL1 mRNA transcripts at baseline are recommended to confirm the diagnosis of CML.
- Bone marrow cytogenetics with a minimum of 20 metaphases is useful to detect additional chromosomal abnormalities (ACAs) in Ph-positive cells, also known as clonal cytogenetic evolution. If bone marrow evaluation is not feasible, fluorescence in situ hybridization (FISH) on the bone marrow or a peripheral blood specimen with dual probes for BCR and ABL1 genes can be used to confirm the diagnosis of CML.”
- “Quantitative RT-PCR (qPCR) should be done at initial workup to establish the presence of quantifiable BCR::ABL1 mRNA transcripts.”
- “Qualitative RT-PCR or D-FISH should be considered for detecting atypical BCR::ABL1 transcripts if there is discordance between FISH and qPCR results.”59
The NCCN provides the following early treatment response milestones for those with CML:59
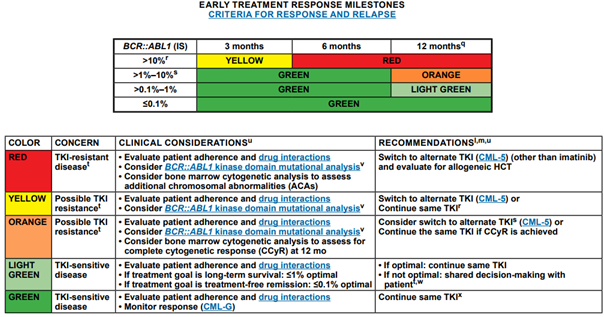
For those discontinuing TKI therapy, the NCCN recommends molecular monitoring every one to two months in the first six months following discontinuation, bimonthly monitoring during months seven to twelve, and quarterly monitoring thereafter (indefinitely) in individuals who remain in MMR (MR3; BCR::ABL1 ≤0.1% IS).59
Acute Lymphoblastic Leukemia (ALL)
Regarding ALL, the NCCN recommends testing of marrow or peripheral blood lymphoblasts using various techniques. The guidelines state the following:
“The diagnosis of ALL generally requires demonstration of ≥20% bone marrow lymphoblasts” predicated “on hematopathology review of bone marrow aspirate and biopsy materials, which includes:
- Morphologic assessment of Wright-Giemsa-stained bone marrow aspirate smears, and hematoxylin and eosin (H&E)-stained core biopsy and clot sections
- Comprehensive flow cytometric immunophenotyping
- Baseline flow cytometric and/or molecular characterization of leukemic clone to facilitate subsequent minimal/measurable residual disease (MRD) analysis
- Karyotyping of G-banded metaphase chromosomes.”60
Regarding molecular characterization of ALL, it is stated that
- “Cytogenetic and molecular prognostic risk stratification for B-cell ALL (B-ALL)
- Optimal risk stratification and treatment planning require testing marrow or peripheral blood lymphoblasts for specific recurrent genetic abnormalities using:
- Interphase fluorescence in situ hybridization (FISH) testing, including probes capable of detecting the major recurrent genetic abnormalities
- Reverse transcriptase polymerase chain reaction (RT-PCR) testing BCR::ABL1 in B-ALL (quantitative or qualitative) including determination of transcript size (ie, p190 vs. p210)
- Comprehensive testing by next-generation sequencing (NGS) for gene fusions and pathogenic mutations is recommended.
- Additional optional tests include:
- Assessment with chromosomal microarray (CMA)/array comparative genomic hybridization (cGH) in cases of aneuploidy or inadequate karyotype.”60
According to the NCCN, “BCR::ABL1–like or Ph–like ALL is a subgroup of B-cell lineage ALL associated with unfavorable prognosis,” and it should be noted that “although this subgroup is Ph-negative, there is an otherwise similar genetic profile to the Ph-positive ALL subgroup including mutation of the IKZF1 gene.” Nevertheless, “this subtype is typically associated with gene fusions and mutations that activate tyrosine kinase pathways as the common mechanism of transformation,” and these gene fusions and mutations include ABL1, ABL2, CRLF2, CSF1R, EPOR, JAK1, JAK2, JAK3, TYK2, PDGFRβ, PDGFRα, FGFR, EBF1, FLT3, IL7R, NTRK3, PTL2B, and SH2B3 genes.”60
As a part of surveillance for individuals with ALL, the NCCN highlights that “an assessment of bone marrow aspirate can be considered as clinically indicated at a frequency of 3 to 6 months for the first 5 years; if a bone marrow aspirate is performed, flow cytometry with additional studies that may include comprehensive cytogenetics, FISH, molecular tests, and MRD assessments should be carried out. While there is insufficient evidence to guide MRD monitoring for patients with Ph-negative disease following completion of maintenance therapy, the approval of blinatumomab, and potentially future therapies for MRD-positive relapse, may warrant testing in this regard.” The NCCN also states that “sequential MRD assessments should be considered for patients who have achieved a complete molecular remission (undetectable levels). The frequency may be increased if MRD levels are detectable or for those discontinuing TKI.”59,60
Pediatric Acute Lymphoblastic Leukemia (PEDALL)
The NCCN also produces guidelines focusing on Pediatric Acute Lymphoblastic Leukemia (PEDALL). For PEDALL, the NCCN provides the following recommendation for genetic characterization.
“GENETIC CHARACTERIZATION
Optimal risk stratification and treatment planning require testing marrow or peripheral blood lymphoblasts for specific recurrent genetic abnormalities using:
- Karyotyping of G-banded metaphase chromosomes
- Interphase fluorescence in situ hybridization (FISH) testing, including probes capable of detecting the major recurrent genetic abnormalities
- Reverse transcriptase-polymerase chain reaction (RT-PCR) testing for BCR::ABL1 in B-ALL (quantitative or qualitative) including determination of transcript size (i.e., p190 vs. p210)
- If BCR::ABL1 negative: encourage testing for gene fusions and mutations associated with BCR::ABL1–like (Ph-like) ALL
- Assessment of various potentially actionable or prognostic mutations and gene fusions via next-generation sequencing (NGS) or alternative methods
- Additional optional tests include:
- Additional assessment (eg, microarray comparative genomic hybridization [CGH] and/or NGS) in cases of aneuploidy or inadequate karyotype
- Whole transcriptome sequencing to identify B-lymphoblastic leukemia/lymphoma (B-ALL/LL) subtypes defined by gene expression profile (ie, ETV6::RUNX1-like, PAX5alt, MYCr).”61
As a part of surveillance under their “Procedures and Molecular Testing” of individuals with PEDALL, the NCCN recommends testing of “bone marrow aspirate and cerebrospinal fluid (CSF) for suspected relapse.” They note that “if bone marrow aspirate is done: Flow cytometry with additional studies that may include comprehensive cytogenetics, FISH, molecular testing, and MRD testing” and suggest that one can “consider periodic BCR::ABL1 transcript-specific quantification (BCR::ABL1+ ALL).”61
The guidelines also state that “the BCR-ALB1-¬like phenotype is associated with recurrent gene fusions and mutations that activate tyrosine kinase pathways and includes gene fusions involving ABL1, ABL2, CRLF2, CSF1R, EPOR, JAK2 or PDGFRB and mutations involving CRLF2, FLT3, IL7R, SH2B3, JAK1, JAK3, and JAK2 (in combination with CRLF2 gene functions). Testing of these abnormalities at diagnosis may aid in risk stratification.”61
European LeukemiaNet (ELN) Working Party
In 2022, an international expert panel on behalf of the ELN published recommendations for the diagnosis and management of AML.
The ELN remarks that “germline predisposition risk should be considered for all patients diagnosed with a hematopoietic malignancy regardless of age, because some germline predisposition alleles, like those in DDX41, can drive hematopoietic malignancies in older age” and notes key features of clinical presentation warranting consideration of clinical testing for a germline predisposition allele(s), including:
“Personal history of ≥ two cancers, one of which is a hematopoietic malignancy (order does not matter)
Personal history of a hematopoietic malignancy plus:
- Another relative within two generations with another hematopoietic malignancy, or
- Another relative within two generations with a solid tumor diagnosed at age 50 or younger, or
- Another relative within two generations with other hematopoietic abnormalities
Presence of a deleterious gene variant in tumor profiling that could be a germline allele, especially if that variant is present during remission*
Age of diagnosis of hematopoietic malignancy at an earlier age than average (eg, MDS diagnosed ≤ 40 y)
Germline status of a variant is confirmed by:
Its presence in DNA derived from a tissue source not likely to undergo somatic mutation frequently (eg, cultured skin fibroblasts or hair follicles) AND at a variant allele frequency consistent with the germline (generally considered between 30-60%), or
Its presence in at least two relatives at a variant allele frequency consistent with the germline.”62 It is also noted that “certain gene alleles (eg, CHEK2 I200T and truncating DDX41 variants) are overwhelmingly likely to be germline and should prompt consideration of germline testing when identified even once.”62
Furthermore, ELN lists tests and procedures recommended to be performed at diagnosis for a patient with AML in the Table 4, captured below.62
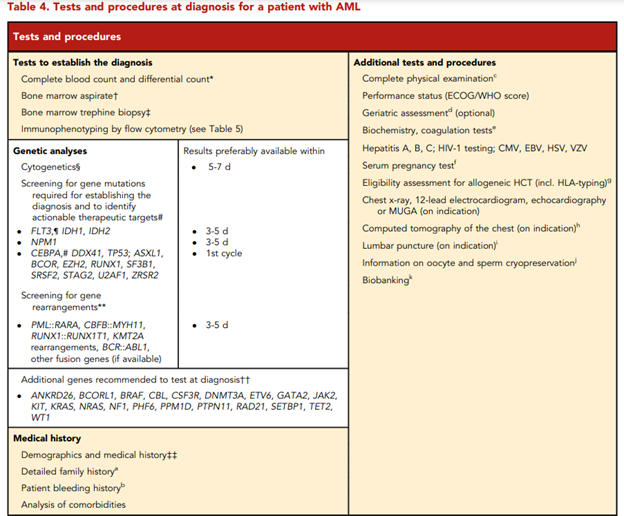
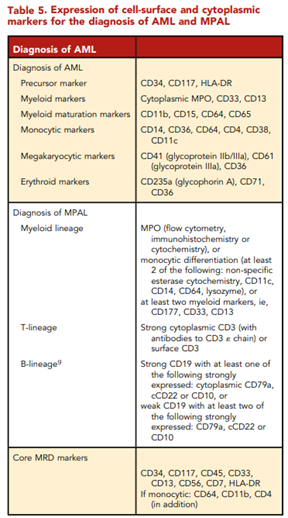
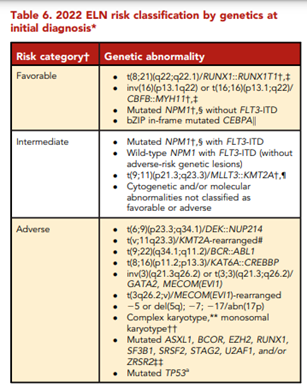
According to ELN, “conventional cytogenetic analysis is mandatory in the evaluation of AML. If conventional cytogenetics fails, fluorescence in situ hybridization is an alternative to detect specific abnormalities like RUNX1::RUNX1T1, CBFB::MYH11, KMT2A (MLL), and MECOM (EVI1) gene fusions, or myelodysplasia-related chromosome abnormalities, eg, loss of chromosome 5q, 7q, or 17p material.” ELN also asserts that “molecular genetic testing should screen for all the genetic abnormalities that define disease and risk categories or that are needed for targeted treatment modalities” listed above in their Table 4, and that for “patients with mutant NPM1 and core-binding factor (CBF)- AML, it is recommended to perform baseline molecular assessment by quantitative polymerase chain reaction (qPCR) or droplet digital PCR (dPCR) to facilitate MRD monitoring after treatment.”62 ELN recognizes that “immunophenotyping by multiparameter flow cytometry (MFC) is required to diagnose AML accurately by identifying cell surface and intracellular markers” and that “it is also important to identify leukemia-associated immunophenotypes (LAIP) for subsequent MRD monitoring by MFC. In cases where an aspirate is unobtainable and circulating blasts are absent, myeloid phenotype may be confirmed on a core biopsy using immunohistochemistry.”62 Cell-surface and cytoplasmic markers of interest for the diagnosis of AML and MPAL as reported by ELN is shown below (Table 5). Lastly, this ELN special report included an updated table of risk classifications at initial diagnosis based on new data (Table 6).
An ELN expert panel has released guidelines for the assessment of measurable residual disease (MRD) of AML that were updated in 2021. First, for molecular MRD recommendations, “techniques for molecular MRD assessment should reach an LOD [limit of detection] of 10-3 or lower. To achieve this LOD, qPCR, dPCR, or error-corrected NGS using UMIs [unique molecular identifiers] is recommended (Grade of Recommendation: B).”
Other recommendations for molecular MRD testing include:
- “For patients with mutant NPM1, CBF AML (RUNX1-RUNX1T1 or CBFB-MYH11), or APL (PML-RARA), we recommend molecular MRD assessment by qPCR or dPCR. (Grade of Recommendation: A)”
- “Leukemia-specific PCR assays (eg, for NPM1, PML-RARA, or CBF AML) are preferred over fewer specific markers, such as WT1 or EV11 expression (Grade of Recommendation: B)”
- “Targeted NGS-MRD using specific mutations identified at diagnosis vs agnostic panel approaches has different strengths and limitations, but both approaches can be considered, depending on sensitivity, turnaround time, resource use, setting (research, clinical trial, or clinical routine), and ability to standardize methodology and reporting. (Grade of Recommendation: B)”
- “If a panel approach is used for NGS-MRD, emerging variants not found at diagnosis should be reported only if confidently detected above background noise. (Grade of Recommendation: B)”
- “For NGS-MRD, we recommend considering all detected mutations as potential MRD markers, with the limitations detailed in recommendations B9 to B11 (listed below) (Grade of Recommendation: B)”
- “Germline mutations (VAF of ∼50 in genes ANKRD26, CEBPA, DDX41, ETV6, GATA2, RUNX1, and TP53) should be excluded as NGS-MRD markers, as they are noninformative for MRD. (Grade of Recommendation: A)”
- “Mutations in DNMT3A, TET2, and ASXL1 (DTA) can be found in age-related clonal hematopoiesis and should be excluded from MRD analysis. (Grade of Recommendation: A)”
- “Mutations in signaling pathway genes (FLT3-ITD, FLT3-TKD, KIT, and RAS, among others) most likely represent residual AML when detected, but are often subclonal and have a low negative predictive value. These mutations are best used in combination with additional MRD markers. (Grade of Recommendation: B).”63
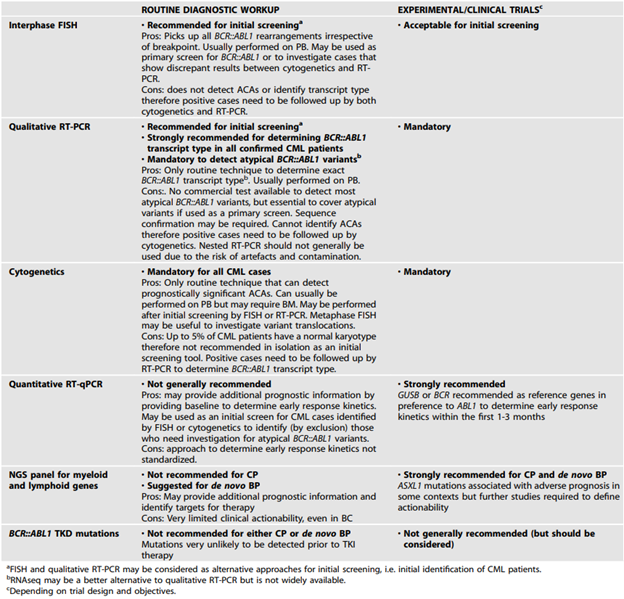
In 2023, ELN published recommendations for the diagnosis and management of CML. In the guidelines they recommend:
- “Cytogenetics along with FISH and/or RT-PCR should be used in all cases to confirm a diagnosis of CML. The limitations of each approach as standalone tests need to be understood and, where appropriate, included in clinical reports.
- Cytogenetic testing should include a screen for ACAs at diagnosis.
- BCR::ABL1 mRNA transcript type should be determined for all cases prior to treatment to enable appropriate follow up.
- The possibility of a rare BCR::ABL1 variant should be excluded. If testing for rare variants is not available, the diagnostic report should clearly state that the presence of a BCR::ABL1 remains a possibility and that further testing in an appropriate reference laboratory should be performed.”64
The ELN also added the following tables with recommended tests for diagnostic workup of CML patients and for monitoring or investigating CML patients on treatment:64

International Workshop on Chronic Lymphocytic Leukemia (iwCLL)
In the iwCLL guidelines for the diagnosis, indications for treatment, response assessment, and supportive management of CLL, the iwCLL reference the World Health Organization in describing “CLL as leukemic, lymphocytic lymphoma, being only distinguishable from small lymphocytic lymphoma (SLL) by its leukemic manifestation” and adduce that “CLL, by definition, is always a disease of neoplastic B cells, whereas the entity formerly described as T-CLL is now called T-cell prolymphocytic leukemia.”65 The iwCLL states that “The diagnosis of CLL requires the presence of ≥5 × 109/L B lymphocytes in the peripheral blood, sustained for at least 3 months. The clonality of these B lymphocytes needs to be confirmed by demonstrating immunoglobulin light chain restriction using flow cytometry.” They also assert that “CLL or SLL might be suspected in otherwise healthy adults who have an absolute increase in clonal B lymphocytes, but who have <5 × 109/L B lymphocytes in the blood. However, in the absence of lymphadenopathy or organomegaly (as detected by physical examination or imaging studies), or of disease-related cytopenias or symptoms, the presence of <5 × 109/L B lymphocytes is defined as monoclonal B lymphocytosis (MBL),” explaining that “The presence of a cytopenia caused by a typical marrow infiltrate establishes the diagnosis of CLL regardless of the number of peripheral blood B lymphocytes or of the lymph node involvement.” Meanwhile, “The definition of SLL requires the presence of lymphadenopathy and the absence of cytopenias caused by a clonal marrow infiltrate. Additionally, the number of B lymphocytes in the peripheral blood should be <5 × 109/L. In SLL, the diagnosis should be confirmed by histopathological evaluation of a lymph node biopsy or biopsy of other tissues.”65
They also mention that “it is important to verify that the patient has CLL and not some other lymphoproliferative disease that can masquerade as CLL, such as hairy cell leukemia or leukemic manifestations of mantle cell lymphoma, marginal zone lymphoma, splenic marginal zone lymphoma with circulating villous lymphocytes, or follicular lymphoma. To achieve this, it is necessary to evaluate the blood smear, the immunophenotype, and, in some cases, the genetic features of the circulating lymphoid cells.”65
British Society for Haematology (BSH)
The Haemato-oncology Task Force of BCSH (British Committee for Standards in Haematology) had produced a draft outline of guidelines for the diagnosis and management of AML in pregnancy, which was subsequently reviewed by the British Society of Haematology due to a consultation process resulting in the name change from BCSH to BSH in 2016. These guidelines state that “as for non-pregnant patients, acute myeloid leukaemia (AML) should be diagnosed using the World Health Organization (WHO) classification (Grade 1A)” and [w]here a diagnosis of leukemia is suspected, care must be taken to ensure that marrow samples are directed for immunophenotypic, cytogenetic and molecular analysis to allow accurate sub-typing and understanding of prognostic features.”66
On the management of older patients with frailty and acute myeloid leukemia, BSH asserts that “older adults who are suspected of having AML and are deemed to be fit to receive anti-leukaemia therapy should undergo the same diagnostic workup as any other patient (Table 1).”67
At diagnosis, “patients should have a bone marrow examination (aspirate and trephine biopsy) for blast enumeration [with a 300 (or 500 where indicated) differential cell count]” and “flow cytometry to characterise the leukaemic clone immunophenotype to confirm lineage, and because this provides a means of assessing measurable residual disease (MRD) in the absence of a molecular marker.” Moreover, “for patients with a high white blood count at presentation, diagnostic workup may be performed on the peripheral blood in lieu of a bone marrow examination, and this may also be a suitable approach for frail patients, for whom best supportive care is the most appropriate treatment option. Consideration should also be given (with appropriate patient consent) to taking trial samples at the time of diagnostic marrow sampling.”67
The BSH notes that “risk stratification remains informed by cytogenetic analysis, and, given that some treatments (e.g., CPX-351) are currently only licensed in the UK for therapy-related AML or AML with myelodysplasia-related changes (AML-MRC), knowledge of cytogenetic abnormalities is a prerequisite to offer the most appropriate treatment.” But while “this is likely to be most efficiently achieved using a multi-target next-generation sequencing (NGS) panel approach with a maximum turnaround time of 21 days,” “we [BSH] recommended rapid screening (turnaround time within 72 hours) for mutations of NPM1, FLT3, IDH1/2 and TP53, given that the presence of NPM1 and IDH1/2 mutations may inform treatment decisions, as these mutations have been shown to confer a superior response to venetoclax/azacitidine), whilst TP53 mutations confer resistance to chemotherapy and an overall poor outcome.67
The BSH also recommends that “at relapse, patients who remain fit to receive anti-leukaemia therapy, should be re-screened for actionable/targetable mutations. At the least, this should include re-screening for mutations in FLT3 (both internal tandem duplication; ITD and tyrosine kinase domain; TKD), given the availability of gilteritinib,” along with “IDH1/2 mutations (if trial recruitment is an option). It should be noted that FLT3 mutations may be acquired or lost at relapse.” As such,
- All patients should have their disease assessed by morphology, immunophenotyping, cytogenetics and molecular studies at presentation.
- At relapse, patients should be re-screened for FLT3 mutations as a minimum.
- Peripheral blood is a suitable alternative to bone marrow for the diagnostic evaluations if there are circulating blast cells.
- All cases should be discussed at an appropriate local or regional haemato-oncology MDT.67
Table 1, produced by BSH and referenced earlier, is captured below:67
In a paper on best practices for laboratory testing of UK patients with AML, BSH recommends that “for a suspected acute leukaemia, most multiparametric flow cytometry laboratories apply a two-stage diagnostic panelan acute leukaemia screen (such as the Euroflow A LOT combination)” in order to confirm leukaemic blasts and allow lineage assessment, “followed immediately by an AML-specific panel if appropriate.” Furthermore, “extended secondary testing may be required to diagnose blastic plasmacytoid dendritic cell neoplasm (BPDCN) or ambiguous leukaemias or acute megakaryoblastic leukaemia.”68
European Society for Medical Oncology (ESMO)
The ESMO released guidelines for the diagnosis of acute myeloid leukemia in adult patients. They recommend molecular testing for c-KIT, FLT3-ITD, FLT3-TKD, NPM1, CEBPA, IDH1, IDH2, and TP53 mutations. They state that “next-generation sequencing (NGS) of a panel of genes commonly mutated in AML provides important additional prognostic and therapeutic information.”69
Acute Lymphoblastic Leukemia (ALL)
The ESMO also published clinical practice guidelines for ALL in 2016 and note that “standard cytogenetics/FISH and especially RT-PCR are routinely performed to obtain a rapid diagnosis of Ph+ ALL and identify certain intermediate/high- and high-risk karyotypes or gene, mainly:
- t(4;11)(q21;q23)/MLL-AFA4, abn11q23/MLL, t(1;19)(q23; p13)/PBX-E2A, t(8;14) or other abn14q32 in non-Burkitt ALL
- del(6q), del(7p), del(17p), −7, +8, low hypodiploidy, i.e., with 30–39 chromosomes/near triploidy with 60–78 chromosomes
- complex (≥5 unrelated clonal abnormalities), and
- T-ALL lacking NOTCH1/FBXW7 mutations and/or with RAS/PTEN abnormalities
The more prognostically favorable cytogenetic/genetic subsets are t(12;21)( p13;q22)/TEL-AML1 + ALL (rare in adults) and hyperdiploid ALL, and NOTCH-1/FBXW7-mutated T-ALL.”70
Chronic Lymphocytic Leukemia
According to ESMO, “the diagnosis of CLL is established by the following criteria:
- Presence of ≥5 × 109/l monoclonal B lymphocytes in the peripheral blood. The clonality of the circulating B lymphocytes needs to be confirmed by demonstrating light chain restriction using flow cytometry.
- The leukaemia cells found in the blood smear are characteristically small, mature-appearing lymphocytes with a narrow border of cytoplasm and a dense nucleus lacking discernible nucleoli and having partially aggregated chromatin. Larger, atypical lymphocytes or prolymphocytes may be seen but must not exceed 55%.”71
Also worth noting is that “CLL cells co-express the B-cell surface antigens CD19 and CD20 together with CD5, CD23, CD43 and CD200. The levels of surface CD20, surface immunoglobulin (Ig) and CD79b are characteristically low compared with those found on normal B cells” and that “each clone of leukaemia cells is restricted to expression of either kappa or lambda Ig light chains, or has no apparent expression of either of the two.” ESMO also states that “Other lymphoma entities to be differentiated from CLL are mantle cell lymphoma (MCL), leukaemic marginal zone lymphoma (MZL) (in particular the splenic variant) and lymphoplasmacytic lymphoma.” This differentiation can be made as “tumour cells may express B-cell surface antigens and CD5, but in most cases, they do not express CD23, in particular MZL. For cases that express CD23, reverse transcription polymerase chain reaction (RT-PCR) for determination of cyclin D1 overexpression, and FISH for detecting a translocation (11;14), but also CD200 expression, are useful for establishing the diagnosis of MCL.” Moreover, “SOX11 staining may be used on tumour biopsies. A diagnosis of MZL is supported by negative or low CD43 expression and high expression of CD180.”71
Ultimately, the ESMO proposes two recommendations regarding the diagnosis of CLL:
- “Diagnosis is usually possible by immunophenotyping of peripheral blood only [III, A].
- LN biopsy and/or bone marrow biopsy may be helpful if immunophenotyping is not conclusive for the diagnosis of CLL [IV, A]."71
During diagnostic and staging work-up, the ESMO recommends also that
- “Binet and Rai staging systems with clinical symptoms are relevant for treatment indication [III, A].
- del(17p), TP53 mutations and IGHV status are relevant for choice of therapy and should be assessed before treatment [III, A].
- Routine evaluation of del(17p), TP53 mutation and IGHV status in early and asymptomatic stage is not recommended [V, D].
- Routine imaging during a watch-and-wait period is not recommended unless there are clinical symptoms [V, E].”71
The ESMO also recommends that “after the first year, when all patients should be seen at 3-monthly intervals, patients can be followed every 3-12 months depending on burden and dynamics of the disease” using history and physical examinations including a careful palpation of all LN areas, spleen and liver and complete blood cell count and differential count.71
Chronic Myeloid Leukemia
The ESMO has published guidelines for diagnosis, treatment, and follow-up of CML. These guidelines include a table which is shown below:
Table 2: Recommendations for diagnostic work-up, assessment of response and monitoring72
| Test |
Baseline (diagnostic work-up) |
To assess the response |
To monitor the response and the treatment |
| Blood counts and differential |
Yes |
Every 15 days until a CHR [complete hematological response] without significant cytopaenias has been achieved |
Every 3 months |
| BM [bone marrow], cytology |
Yes |
No |
No |
| BM, karyotype |
Yes |
At 3 and 6 months |
Then every 6 months until CCyR [complete cytogenetic response] has been achieved |
| Blood, iFISH |
No |
No |
Only if cytogenetics of BM metaphases cannot be analyzed or is normal and molecular response cannot be assessed |
| Blood, RT-PCR (qualitative) |
Yes |
No |
No |
| Blood, qRT-PCR (quantitative, BCR–ABL %) |
No |
Every 3 months |
Every 4–6 weeks in first year after treatment discontinuation |
| Mutational Analysis |
Only in AP [accelerated phase] or BP [blast phase] |
No |
Only in the case of failure |
The ESMO has also stated that a “diagnosis must be confirmed by cytogenetics showing t(9;22)(q34;q11), and by multiplex RT-PCR showing BCR–ABL1 transcripts.”72 Other warning signs include “major route cytogenetic aberrations (+8, iso(17q), +19, +22q-), chromosome 3 aberrations and BM fibrosis at diagnosis signs”; further, a quantification of BCR-ABL mRNA is required every three months.72 Finally, the ESMO acknowledges that mutation analysis is “due” in case of failure of first-line therapy or if BCR-ABL transcript levels increase. However, ESMO recommends against baseline mutational analysis in patients with newly diagnosed CML-CP.72
References
- ACCC. Hematologic Malignancies. https://www.accc-cancer.org/home/learn/cancer-types/hematologic-malignancies
- NCBI. Myeloid Neoplasm. https://www.ncbi.nlm.nih.gov/medgen/445430
- NCBI. Lymphoid Neoplasm. https://www.ncbi.nlm.nih.gov/medgen/108626
- Iacobucci I, Mullighan CG. Genetic Basis of Acute Lymphoblastic Leukemia. J Clin Oncol. Mar 20 2017;35(9):975-983. doi:10.1200/jco.2016.70.7836
- Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA: a cancer journal for clinicians. Jan 2017;67(1):7-30. doi:10.3322/caac.21387
- Yamamoto JF, Goodman MT. Patterns of leukemia incidence in the United States by subtype and demographic characteristics, 1997-2002. Cancer causes & control : CCC. May 2008;19(4):379-90. doi:10.1007/s10552-007-9097-2
- Schiffer CA, Gurbuxani S. Acute myeloid leukemia: Clinical manifestations, pathologic features, and diagnosis. Updated June 19, 2024. https://www.uptodate.com/contents/acute-myeloid-leukemia-clinical-manifestations-pathologic-features-and-diagnosis
- CGARN. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England journal of medicine. May 30 2013;368(22):2059-74. doi:10.1056/NEJMoa1301689
- Stock W, Thirman MJ. Acute myeloid leukemia: Molecular genetics. Updated November 13, 2024. https://www.uptodate.com/contents/acute-myeloid-leukemia-molecular-genetics
- Falini B, Mecucci C, Tiacci E, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. The New England journal of medicine. Jan 20 2005;352(3):254-66. doi:10.1056/NEJMoa041974
- Schiffer C, Uy G. Acute myeloid leukemia: Risk factors and prognosis. Updated January 15, 2025. https://www.uptodate.com/contents/acute-myeloid-leukemia-risk-factors-and-prognosis
- Castells MC, Akin C. Mastocytosis (cutaneous and systemic) in adults: Epidemiology, pathogenesis, clinical manifestations, and diagnosis. UpToDate. Updated July 11, 2024. https://www.uptodate.com/contents/mastocytosis-cutaneous-and-systemic-in-adults-epidemiology-pathogenesis-clinical-manifestations-and-diagnosis
- Yui S, Kurosawa S, Yamaguchi H, et al. D816 mutation of the KIT gene in core binding factor acute myeloid leukemia is associated with poorer prognosis than other KIT gene mutations. Ann Hematol. Oct 2017;96(10):1641-1652. doi:10.1007/s00277-017-3074-y
- Hou HA, Huang TC, Lin LI, et al. WT1 mutation in 470 adult patients with acute myeloid leukemia: stability during disease evolution and implication of its incorporation into a survival scoring system. Blood. Jun 24 2010;115(25):5222-31. doi:10.1182/blood-2009-12-259390
- Vosberg S, Hartmann L, Metzeler KH, et al. Relapse of acute myeloid leukemia after allogeneic stem cell transplantation is associated with gain of WT1 alterations and high mutation load. Haematologica. Dec 2018;103(12):e581-e584. doi:10.3324/haematol.2018.193102
- Rai KR, Stilgenbauer S. Pathobiology of chronic lymphocytic leukemia. Updated June 27, 2025. https://www.uptodate.com/contents/pathobiology-of-chronic-lymphocytic-leukemia
- Frucht H, Lucas AL. Molecular genetics of colorectal cancer. UpToDate. Updated November 20, 2024. https://www.uptodate.com/contents/molecular-genetics-of-colorectal-cancer
- Rai KR, Stilgenbauer S, Aster JC. Clinical features and diagnosis of chronic lymphocytic leukemia/small lymphocytic lymphoma. Updated May 30, 2025. https://www.uptodate.com/contents/clinical-features-and-diagnosis-of-chronic-lymphocytic-leukemia-small-lymphocytic-lymphoma
- Amin NA, Malek SN. Gene mutations in chronic lymphocytic leukemia. Semin Oncol. Apr 2016;43(2):215-21. doi:10.1053/j.seminoncol.2016.02.002
- Nowell P, Hungerford D. A minute chromosome in human granulocytic leukemia. Science. 1960-11-18 1960:1497. doi:10.1126/science.132.3438.1488
- Schrijver I, Zehnder JL. Genetic abnormalities in hematologic and lymphoid malignancies. Updated June 5, 2024. https://www.uptodate.com/contents/genetic-abnormalities-in-hematologic-and-lymphoid-malignancies
- Kurzrock R, Kantarjian HM, Druker BJ, Talpaz M. Philadelphia chromosome-positive leukemias: from basic mechanisms to molecular therapeutics. Annals of internal medicine. 2003;138(10):819-30. http://thinkkimi.com/Work/philadelphia.pdf
- Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nature reviews Cancer. Mar 2005;5(3):172-83. doi:10.1038/nrc1567
- Warmuth M, Danhauser-Riedl S, Hallek M. Molecular pathogenesis of chronic myeloid leukemia: implications for new therapeutic strategies. Ann Hematol. Feb 1999;78(2):49-64. doi:10.1007/s002770050473
- Van Etten RA. Chronic myeloid leukemia: Pathogenesis, clinical manifestations, and diagnosis. Updated April 23, 2025. https://www.uptodate.com/contents/chronic-myeloid-leukemia-pathogenesis-clinical-manifestations-and-diagnosis
- Leoni V, Biondi A. Tyrosine kinase inhibitors in BCR-ABL positive acute lymphoblastic leukemia. Haematologica. 2015;100(3):295-9. doi:10.3324/haematol.2015.124016
- Tachibana T, Kondo T, Uchida N, et al. The clinical significance of BCR-ABL1 mutations in patients with Philadelphia chromosome-positive chronic myeloid leukemia who underwent allogeneic hematopoietic cell transplantation. Transplantation and Cellular Therapy. 2022/03/13/ 2022;doi:10.1016/j.jtct.2022.03.009
- PDQ Adult Treatment Editorial Board. Adult Acute Lymphoblastic Leukemia Treatment (PDQ®). 2025. https://www.ncbi.nlm.nih.gov/books/NBK65727/
- Pui CH. Recent advances in acute lymphoblastic leukemia. Oncology (Williston Park, NY). Apr 15 2011;25(4):341, 346-7.
- Leonard DGB. Molecular Pathology in Clinical Practice. Springer; 2016. http://www.springer.com/us/book/9783319196732
- Ampasavate C, Jutapakdee W, Phongpradist R, et al. FLT3, a prognostic biomarker for acute myeloid leukemia (AML): Quantitative monitoring with a simple anti-FLT3 interaction and flow cytometric method. J Clin Lab Anal. May 2019;33(4):e22859. doi:10.1002/jcla.22859
- Alonso CM, Llop M, Sargas C, et al. Clinical Utility of a Next-Generation Sequencing Panel for Acute Myeloid Leukemia Diagnostics. J Mol Diagn. Mar 2019;21(2):228-240. doi:10.1016/j.jmoldx.2018.09.009
- Yeung CC, Egan D, Radich JP. Molecular monitoring of chronic myeloid leukemia: present and future. Expert review of molecular diagnostics. Oct 2016;16(10):1083-1091. doi:10.1080/14737159.2016.1227243
- FDA. Evaluation of Automatic Class III Designation for QuantideX qPCR BCR-ABL1 IS Kit Decision Summary. https://www.accessdata.fda.gov/cdrh_docs/reviews/DEN160003.pdf
- Brown JT, Beldorth IJ, Laosinchai-Wolf W, et al. Analytical Validation of a Highly Sensitive, Multiplexed Chronic Myeloid Leukemia Monitoring System Targeting BCR-ABL1 RNA. The Journal of molecular diagnostics : JMD. 2019;21(4):718-733. doi:10.10162Fj.jmoldx.2019.03.002
- DeZern AE, Sung A, Kim S, et al. Role of allogeneic transplantation for FLT3/ITD acute myeloid leukemia: outcomes from 133 consecutive newly diagnosed patients from a single institution. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. Sep 2011;17(9):1404-9. doi:10.1016/j.bbmt.2011.02.003
- Pastore F, Greif PA, Schneider S, et al. The NPM1 mutation type has no impact on survival in cytogenetically normal AML. PloS one. 2014;9(10):e109759. doi:10.1371/journal.pone.0109759
- Willemze R, Suciu S, Meloni G, et al. High-dose cytarabine in induction treatment improves the outcome of adult patients younger than age 46 years with acute myeloid leukemia: results of the EORTC-GIMEMA AML-12 trial. J Clin Oncol. Jan 20 2014;32(3):219-28. doi:10.1200/jco.2013.51.8571
- Devillier R, Mansat-De Mas V, Gelsi-Boyer V, et al. Role of ASXL1 and TP53 mutations in the molecular classification and prognosis of acute myeloid leukemias with myelodysplasia-related changes. Oncotarget. Apr 10 2015;6(10):8388-96. doi:10.18632/oncotarget.3460
- Bolouri H, Farrar JE, Triche T, Jr., et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. Jan 2018;24(1):103-112. doi:10.1038/nm.4439
- Jongen-Lavrencic M, Grob T, Hanekamp D, et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. The New England journal of medicine. Mar 29 2018;378(13):1189-1199. doi:10.1056/NEJMoa1716863
- Kuwatsuka Y, Tomizawa D, Kihara R, et al. Prognostic value of genetic mutations in adolescent and young adults with acute myeloid leukemia. International journal of hematology. Feb 2018;107(2):201-210. doi:10.1007/s12185-017-2340-z
- Zhu YM, Wang PP, Huang JY, et al. Gene mutational pattern and expression level in 560 acute myeloid leukemia patients and their clinical relevance. Journal of translational medicine. Aug 22 2017;15(1):178. doi:10.1186/s12967-017-1279-4
- Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. The New England journal of medicine. Jun 9 2016;374(23):2209-2221. doi:10.1056/NEJMoa1516192
- Sperr WR, Zach O, Poll I, et al. Karyotype plus NPM1 mutation status defines a group of elderly patients with AML (>/=60 years) who benefit from intensive post-induction consolidation therapy. American journal of hematology. Dec 2016;91(12):1239-1245. doi:10.1002/ajh.24560
- Heiblig M, Sujobert P, Hayette S, et al. Impact of NPM1 mutation subtypes on treatment outcome in AML: The Lyon-University Hospital experience. Leukemia research. Jan 2019;76:29-32. doi:10.1016/j.leukres.2018.11.016
- D’Adda M, Farina M, Schieppati F, et al. The e13a2 BCR-ABL transcript negatively affects sustained deep molecular response and the achievement of treatment-free remission in patients with chronic myeloid leukemia who receive tyrosine kinase inhibitors. Cancer. 2019/02/01 2019;0(0)doi:10.1002/cncr.31977
- Greenfield G, McMullan R, Robson N, McGimpsey J, Catherwood M, McMullin MF. Response to Imatinib therapy is inferior for e13a2 BCR-ABL1 transcript type in comparison to e14a2 transcript type in chronic myeloid leukaemia. BMC Hematol. 2019;19:7. doi:10.1186/s12878-019-0139-2
- Xu J, Zhang Y, Hu J, Ren Y, Wang H. Clinical features and prognosis of normal karyotype acute myeloid leukemia pediatric patients with WT1 mutations: an analysis based on TCGA database. Hematology. Dec 2020;25(1):79-84. doi:10.1080/16078454.2020.1720102
- Sasaki K, Kanagal-Shamanna R, Montalban-Bravo G, et al. Impact of the variant allele frequency of ASXL1, DNMT3A, JAK2, TET2, TP53, and NPM1 on the outcomes of patients with newly diagnosed acute myeloid leukemia. Cancer. Feb 15 2020;126(4):765-774. doi:10.1002/cncr.32566
- Duncavage EJ, Schroeder MC, O'Laughlin M, et al. Genome Sequencing as an Alternative to Cytogenetic Analysis in Myeloid Cancers. The New England journal of medicine. Mar 11 2021;384(10):924-935. doi:10.1056/NEJMoa2024534
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. May 19 2016;127(20):2391-405. doi:10.1182/blood-2016-03-643544
- Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. Jul 2022;36(7):1703-1719. doi:10.1038/s41375-022-01613-1
- Li W. The 5(th) Edition of the World Health Organization Classification of Hematolymphoid Tumors. In: Li W, ed. Leukemia. Exon Publications; 2022.
- de Haas V, Ismaila N, Zhang L. Initial Diagnostic Workup of Acute Leukemia: ASCO Clinical Practice Guideline Endorsement Summary of the CAP and ASH Guideline. J Oncol Pract. Feb 2019;15(2):101-105. doi:10.1200/jop.18.00613
- Arber DA, Borowitz MJ, Cessna M, et al. Initial Diagnostic Workup of Acute Leukemia: Guideline From the College of American Pathologists and the American Society of Hematology. Arch Pathol Lab Med. Oct 2017;141(10):1342-1393. doi:10.5858/arpa.2016-0504-CP
- NCCN. NCCN Clinical Practice Guidelines in Oncology; Acute Myeloid Leukemia Version 2.2025. Updated January 27, 2025. https://www.nccn.org/professionals/physician_gls/pdf/aml.pdf
- NCCN. Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma Version 3.2025. Updated April 2, 2025. https://www.nccn.org/professionals/physician_gls/pdf/cll.pdf
- NCCN. NCCN Clinical Practice Guidelines in Oncology: Chronic Myeloid Leukemia Version 1.2026. Updated July 16, 2025. https://www.nccn.org/professionals/physician_gls/pdf/cml.pdf
- NCCN. NCCN Clinical Practice Guidelines in Oncology: Acute Lymphoblastic Leukemia Version 2.2025. Updated June 27, 2025. https://www.nccn.org/professionals/physician_gls/pdf/all.pdf
- NCCN. NCCN Clinical Practice Guidelines in Oncology Pediatric Acute Lymphoblastic Leukemia Version 3.2025. National Comprehensive Cancer Network. Updated March 17 2025. https://www.nccn.org/professionals/physician_gls/pdf/ped_all.pdf
- Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. Sep 22 2022;140(12):1345-1377. doi:10.1182/blood.2022016867
- Heuser M, Freeman SD, Ossenkoppele GJ, et al. 2021 Update on MRD in acute myeloid leukemia: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2021;138(26):2753-2767. doi:10.1182/blood.2021013626
- Cross NCP, Ernst T, Branford S, et al. European LeukemiaNet laboratory recommendations for the diagnosis and management of chronic myeloid leukemia. Leukemia. 2023/11/01 2023;37(11):2150-2167. doi:10.1038/s41375-023-02048-y
- Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. Jun 21 2018;131(25):2745-2760. doi:10.1182/blood-2017-09-806398
- Ali S, Jones GL, Culligan DJ, et al. Guidelines for the diagnosis and management of acute myeloid leukaemia in pregnancy. Br J Haematol. Aug 2015;170(4):487-95. doi:10.1111/bjh.13554
- Dennis M, Copland M, Kaur H, et al. Management of older patients with frailty and acute myeloid leukaemia: A British Society for Haematology good practice paper. Br J Haematol. Oct 2022;199(2):205-221. doi:10.1111/bjh.18369
- Mehta P, Telford N, Wragg C, et al. Recommendations for laboratory testing of UK patients with acute myeloid leukaemia. Br J Haematol. Jan 2023;200(2):150-159. doi:10.1111/bjh.18516
- Heuser M, Ofran Y, Boissel N, et al. Acute myeloid leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. Jun 2020;31(6):697-712. doi:10.1016/j.annonc.2020.02.018
- Hoelzer D, Bassan R, Dombret H, Fielding J, Ribera M, Buske C. ESMO Consensus Guidelines for the Management of Patients with Metastatic Colorectal Cancer. 2016;doi:10.1093/annonc/mdw235
- Eichhorst B, Robak T, Montserrat E, et al. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. Jan 2021;32(1):23-33. doi:10.1016/j.annonc.2020.09.019
- Hochhaus A, Saussele S, Rosti G, et al. Chronic myeloid leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. Jul 1 2017;28(suppl_4):iv41-iv51. doi:10.1093/annonc/mdx219
- FDA. Premarket Approval (PMA) for LeukoStrat CDx FLT3 Mutation Assay. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P160040
- FDA. Premarket Approval Order for Abbott RealTime IDH2. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P170005
Coding Section
| Code | Number |
Description |
| CPT | 81120 |
IDH1 (isocitrate dehydrogenase 1 [NADP+], soluble) (e.g., glioma), common variants (e.g., R132H, R132C) |
| 81121 |
IDH2 (isocitrate dehydrogenase 2 [NADP+], mitochondrial) (e.g., glioma), common variants (e.g., R140W, R172M) |
|
| 81170 |
ABL1 (ABL proto-oncogene 1, non-receptor tyrosine kinase) (e.g., acquired imatinib tyrosine kinase inhibitor resistance), gene analysis, variants in the kinase domain |
|
| 81175 |
ASXL1 (additional sex combs like 1, transcriptional regulator) (e.g., myelodysplastic syndrome, myeloproliferative neoplasms, chronic myelomonocytic leukemia), gene analysis; full gene sequence |
|
| 81176 |
ASXL1 (additional sex combs like 1, transcriptional regulator) (e.g., myelodysplastic syndrome, myeloproliferative neoplasms, chronic myelomonocytic leukemia), gene analysis; targeted sequence analysis (e.g., exon 12) |
|
| 81195 (effective 01/01/2025) | Cytogenomic (genome-wide) analysis, hematologic malignancy, structural variants and copy number variants, optical genome mapping (OGM) | |
| 81206 |
BCR/ABL1 (t(9:22)) (e.g., chronic myelogenous leukemia) translocation analysis; major breakpoint, qualitative or quantitative |
|
| 81207 |
BCR/ABL1 (t(9:22)) (e.g., chronic myelogenous leukemia) translocation analysis; minor breakpoint, qualitative or quantitative |
|
| 81208 |
BCR/ABL1 (t(9:22)) (e.g., chronic myelogenous leukemia) translocation analysis; other breakpoint, qualitative or quantitative |
|
| 81218 |
CEBPA (CCAAT/enhancer binding protein [C/EBP], alpha) (e.g., acute myeloid leukemia), gene analysis, full gene sequence |
|
| 81245 |
FLT3 (fms-related tyrosine kinase 3) (e.g., acute myeloid leukemia), gene analysis; internal tandem duplication (ITD) variants (ie, exons 14, 15) |
|
| 81246 |
FLT3 (fms-related tyrosine kinase 3) (e.g., acute myeloid leukemia), gene analysis; tyrosine kinase domain (TKD) variants (e.g., D835, I836) |
|
| 81261 | IGH@ (Immunoglobulin heavy chain locus) (eg, leukemias and lymphomas, B-cell), gene rearrangement analysis to detect abnormal clonal population(s); amplified methodology (eg, polymerase chain reaction) | |
| 81262 | IGH@ (Immunoglobulin heavy chain locus) (eg, leukemias and lymphomas, B-cell), gene rearrangement analysis to detect abnormal clonal population(s); direct probe methodology (eg, Southern blot) | |
| 81263 | IGH@ (Immunoglobulin heavy chain locus) (eg, leukemia and lymphoma, B-cell), variable region somatic mutation analysis | |
| 81272 |
KIT (v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog) (e.g., gastrointestinal stromal tumor [GIST], acute myeloid leukemia, melanoma), gene analysis, targeted sequence analysis (e.g., exons 8, 11, 13, 17, 18) |
|
| 81273 |
KIT (v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog) (e.g., mastocytosis), gene analysis, D816 variant(s) |
|
| 81310 |
NPM1 (nucleophosmin) (e.g., acute myeloid leukemia) gene analysis, exon 12 variants |
|
| 81334 |
RUNX1 (runt related transcription factor 1) (e.g., acute myeloid leukemia, familial platelet disorder with associated myeloid malignancy) gene analysis, targeted sequence analysis (e.g., exons 3 – 8) |
|
| 81351 | TP53 (tumor protein 53) (eg, Li-Fraumeni syndrome) gene analysis; full gene sequence | |
| 81352 | TP53 (tumor protein 53) (eg, Li-Fraumeni syndrome) gene analysis; targeted sequence analysis (eg, 4 oncology) | |
| 81401 | Molecular pathology procedure, Level 2 (eg, 2-10 SNPs, 1 methylated variant, or 1 somatic variant [typically using nonsequencing target variant analysis], or detection of a dynamic mutation disorder/triplet repeat) | |
| 81403 |
Molecular pathology procedure, Level 4 (e.g., analysis of single exon by DNA sequence analysis, analysis of > 10 amplicons using multiplex PCR in 2 or more independent reactions, mutation scanning or duplication/deletion variants of 2 – 5 exons) |
|
| 81405 |
Molecular pathology procedure, Level 6 (e.g., analysis of 6 – 10 exons by DNA sequence analysis, mutation scanning or duplication/deletion variants of 11 – 25 exons, regionally targeted cytogenomic array analysis) |
|
| 81479 |
Unlisted molecular pathology procedure |
|
| 88237 | Tissue culture for neoplastic disorders; bone marrow, blood cells | |
| 88271 |
Molecular cytogenetics; DNA probe, each (e.g., FISH) |
|
| 88272 |
Molecular cytogenetics; chromosomal in situ hybridization, analyze 3 – 5 cells (e.g., for derivatives and markers) |
|
| 88273 |
Molecular cytogenetics; chromosomal in situ hybridization, analyze 10 – 30 cells (e.g., for microdeletions) |
|
| 88274 |
Molecular cytogenetics; interphase in situ hybridization, analyze 25 – 99 cells |
|
| 88275 |
Molecular cytogenetics; interphase in situ hybridization, analyze 100 – 300 cells |
|
| 88291 | Cytogenetics and molecular cytogenetics, interpretation and report | |
| 0016U |
Oncology (hematolymphoid neoplasia), RNA, BCR/ABL1 major and minor breakpoint fusion transcripts, quantitative PCR amplification, blood or bone marrow, report of fusion not detected or detected with quantitation Lab/Manufacturer: University of Iowa, Department of Pathology, Asuragen |
|
| 0023U |
Oncology (acute myelogenous leukemia), DNA, genotyping of internal tandem duplication, p.D835, p.I836, using mononuclear cells, reported as detection or non-detection of FLT3 mutation and indication for or against the use of midostaurin |
|
| 0040U |
BCR/ABL1 (t(9;22)) (e.g., chronic myelogenous leukemia) translocation analysis, major breakpoint, quantitative Lab/Manufacturer: MolecularMD |
|
| 0046U |
FLT3 (fms-related tyrosine kinase 3) (e.g., acute myeloid leukemia) internal tandem duplication (ITD) variants, quantitative |
|
| 0049U |
NPM1 (nucleophosmin) (e.g., acute myeloid leukemia) gene analysis, quantitative |
|
| 0413U |
Oncology (hematolymphoid neoplasm), optical genome mapping for copy number alterations, aneuploidy, and balanced/complex structural rearrangements, DNA from blood or bone marrow, report of clinically significant alterations |
|
| 0592U (effective 10/01/2025) | Oncology (hematolymphoid neoplasms), DNA, targeted genomic sequence of 417 genes, interrogation for gene fusions, translocations, rearrangements, utilizing formalin-fixed paraffinembedded (FFPE) tumor tissue, results report clinically significant variant(s) |
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community, and other nonaffiliated technology evaluation centers, reference to federal regulations, other plan medical policies, and accredited national guidelines.
"Current Procedural Terminology © American Medical Association. All Rights Reserved"
History From 2023 Forward
| 10/23/2025 | Annual review, no change to policy intent. Updating description, table of terminology, rationale, and references. |
| 09/25/2025 | Adding code 0592U effective 10/01/2025 |
| 01/22/2025 | Added CPT code 81195 effective 01/01/2025 |
| 10/22/2024 | Annual review, policy updated to include BCR-ABL1 testing and new criteria #7 has been added to address optimal risk stratification and treatment planning. Updating note 1 to direct reader to CAM 235. Also updating rationale, references and multiple coding additions. |
| 10/25/2023 |
New Policy |