
Whole Genome and Whole Exome Sequencing - CAM 291
Description
Whole genome sequencing (WGS) is the strategy of using next-generation technology to sequence the entire genome. Whole exome Ssquencing (WES) refers to sequencing of the exome, the component of the genome that predominantly encode proteins. Next generation sequencing (NGS) involves sequencing of multiple small fragments of DNA in parallel, producing fast, accurate sequencing results (Hulick, 2024).
This policy is applicable for undiagnosed rare germline disorders.
Regulatory Status
Genotyping is considered a laboratory developed test (LDT); developed, validated and performed by individual laboratories. Additionally, many labs have developed specific tests that they must validate and perform in house. These laboratory-developed tests (LDTs) are regulated by the Centers for Medicare & Medicaid Services (CMS) as high-complexity tests under the Clinical Laboratory Improvement Amendments of 1988 (CLIA ’88). As an LDT, the U.S. Food and Drug Administration has not approved or cleared this test; however, FDA clearance or approval is not currently required for clinical use.
Policy
Application of coverage criteria is dependent upon an individual’s benefit coverage at the time of the request.
- For the evaluation of unexplained congenital or neurodevelopmental disorder in individuals less than 18 years of age, whole exome sequencing (WES) and comparator analysis (e.g., parents/siblings) WES is considered MEDICALLY NECESSARY when all of the following criteria are met:
- When the individual has been evaluated by an ABMGG board-certified medical geneticist or an ABGC board-certified genetic counselor (CGC) and has been counseled about the potential risks of genetic testing;
- When the WES results will impact patient management and clinical outcome for the individual being tested;
- When a genomic etiology is the most likely explanation for the phenotype;
- When no other causative circumstances (e.g., environmental exposures, injury, infection) can explain the symptoms;
- When the clinical presentation does not fit a well-described syndrome for which single-gene or targeted panel testing (e.g., comparative genomic hybridization/chromosomal microarray analysis) is available;
- When the differential diagnosis list and/or phenotype warrant testing of multiple genes and one of the following:
- WES is more practical than the separate single gene tests or panels that would be recommended based on the differential diagnosis.
- WES results may preclude the need for multiple and/or invasive procedures, follow-up, or screening that would be recommended in the absence of testing.
- For a fetus with ultrasound anomalies, WES and comparator analysis (e.g., parents/siblings) WES is considered MEDICALLY NECESSARY when all of the following criteria are met:
- When pre-test counseling has been provided by an ABMGG board-certified medical geneticist or an ABGC board-certified genetic counselor (CGC);
- When standard chromosomal microarray testing (CMA) and karyotype analysis have failed to yield a definitive diagnosis;
- When a genomic etiology is the most likely explanation for the phenotype;
- When no other causative circumstances (e.g., environmental exposures, injury, infection) can explain the symptoms;
- When clinical presentation does not fit a well-described syndrome for which single-gene or targeted panel testing is available. If a specific diagnosis is suspected, molecular testing for the suggested disorder (with single-gene test or gene panel) should be the initial test.
- Reanalysis of exome sequencing (ES) data with WES and comparator analysis (e.g., parents/siblings) WES is considered MEDICALLY NECESSARY for any of the following situations:
- For individuals less than 18 years of age with initial negative ES results as an aid in clinical diagnosis when additional phenotypic findings are noted during a child’s growth and development.
- For diagnostic results and results deemed to be possibly (but not definitively) associated with the fetal phenotype (new gene-disease associations might have been unknown at the time of initial diagnosis).
- For fetal ES with nondiagnostic or negative results, if a new phenotype develops in the proband after birth, a future pregnancy is planned, or a significant amount of time has passed (at least 12 months) since the initial testing was performed.
- If the original prenatal ES report does not account for the complete phenotype or if new/additional phenotypes develop over time.
- When WES is unable to identify a causative variant and the clinical suspicion of a genomic etiology remains in situations where any of the above criteria are met in their entirety, whole genome sequencing (WGS) MEETS COVERAGE CRITERIA.
The following does not meet coverage criteria due to a lack of available published scientific literature confirming that the test(s) is/are required and beneficial for the diagnosis and treatment of an individual’s illness.
- If WES has been previously performed, further genetic tests involving only exome analyses is considered NOT MEDICALLY NECESSARY.
- Focused exome sequencing and targeted WGS is considered NOT MEDICALLY NECESSARY.
- For all other situations not described above, WES is considered NOT MEDICALLY NECESSARY.
- For all other situations not described above, WGS is considered NOT MEDICALLY NECESSARY.
Table of Terminology
| Term |
Definition |
| AAN |
American Academy of Neurology |
| AANEM |
American Association of Neuromuscular and Electrodiagnostic Medicine |
| AAP |
American Academy of Pediatrics |
| ABGC |
American Board of Genetic Counseling |
| ABMGG |
American Board of Medical Genetics and Genomics |
| ACMG |
American College of Medical Genetics and Genomics |
| ACOG |
American College of Obstetricians and Gynecologists |
| ACTA2 |
Actin alpha 2 smooth muscle |
| ACTC1 |
Actin alpha cardiac muscle 1 |
| AFF2 |
AF4/FMR2 family member 2 |
| AMP |
Association for Molecular Pathology |
| APC |
APC regulator of WNT signalling pathway |
| APOB |
Apolipoprotein B |
| AR |
Androgen receptor |
| ASD |
Autism spectrum disorder |
| ATN1 |
Atrophin 1 |
| ATP7B |
ATPase copper transporting beta |
| ATXN1 |
Ataxin 1 |
| ATXN10 |
Ataxin 10 |
| ATXN2 |
Ataxin 2 |
| ATXN3 |
Ataxin 3 |
| ATXN7 |
Ataxin 7 |
| ATXN8OS |
Ataxin 8 opposite strand lncRNA |
| AXL |
AXL receptor tyrosine kinase |
| BMPR1A |
Bone morphogenetic protein receptor type 1A |
| BRCA1 |
BRCA1 DNA repair associated |
| BRCA2 |
BRCA2 DNA repair associated |
| BTD |
Biotinidase |
| C9orf72 |
C9orf72-SMCR8 complex subunit |
| CA |
Congenital anomalies |
| CACNA1A |
Calcium voltage-gated channel subunit alpha1 A |
| CACNA1S |
Calcium voltage-gated channel subunit alpha1 S |
| CCDC141 |
Coiled-coil domain containing 141 |
| CCDC88C |
Coiled-coil domain containing 88C |
| CDON |
Cell adhesion associated; oncogene regulated |
| CES |
Clinical exome sequencing |
| CFES |
Clinically focused exome sequencing |
| CGC |
ABGC board-certified genetic counselor |
| CHD7 |
Chromodomain helicase DNA binding protein 7 |
| CLIA ’88 |
Clinical Laboratory Improvement Amendments of 1988 |
| CMA |
Chromosomal microarray analysis |
| CMS |
Centers for Medicare & Medicaid Services |
| CNBP |
CCHC-type zinc finger nucleic acid binding protein |
| COL3A1 |
Collagen type III alpha 1 chain |
| CSTB |
Cystatin B |
| DCAF17 |
DDB1 and CUL4 associated factor 17 |
| DCC |
DCC netrin 1 receptor |
| DD |
Developmental delay |
| DIP2B |
Disco interacting protein 2 homolog B |
| DMPK |
DM1 protein kinase |
| DMXL2 |
Dmx like 2 |
| DNA |
Deoxyribonucleic acid |
| DSC2 |
Desmocollin 2 |
| DSG2 |
Desmoglein 2 |
| DSP |
Desmoplakin |
| EGF |
Epidermal growth factor |
| EP |
Expected pathogenic |
| ES |
Exome sequencing |
| FBN1 |
Fibrillin 1 |
| FGFR1 |
Fibroblast growth factor receptor 1 |
| FMR1 |
Fragile X messenger ribonucleoprotein 1 |
| FXN |
Frataxin |
| GADL1 |
Glutamate decarboxylase like 1 |
| GDD |
Global developmental delay |
| Gdna |
Genomic DNA |
| GLA |
Galactosidase alpha |
| GnRH |
Gonadotropin-releasing hormone |
| GS |
Genome sequencing |
| HTT |
Huntingtin |
| ID |
Intellectual disability |
| IGSF10 |
Immunoglobulin superfamily member 10 |
| IHH |
Idiopathic hypogonadotropic hypogonadism |
| ISPD |
International Society for Prenatal Diagnosis |
| JAMA |
Journal of the American Medical Association |
| JPH3 |
Junctophilin 3 |
| KCNH2 |
Potassium voltage-gated channel subfamily H member 2 |
| KCNQ1 |
Potassium voltage-gated channel subfamily Q member 1 |
| KP |
Known pathogenic |
| LDLR |
Low density lipoprotein receptor |
| LDT |
Laboratory developed test |
| LMNA |
Lamin A/C |
| MCC |
MCC regulator of WNT signalling pathway |
| MEN1 |
Menin 1 |
| MLH1 |
MutL homolog 1 |
| MSH2 |
MutS homolog 2 |
| MSH6 |
MutS homolog 6 |
| MUTYH |
MutY DNA glycosylase |
| MYBPC3 |
Myosin binding protein C3 |
| MYH11 |
Myosin heavy chain 11 |
| MYH7 |
Myosin heavy chain 7 |
| MYL2 |
Myosin light chain 2 |
| MYL3 |
Myosin light chain 3 |
| NOA |
Nonobstructive azoospermia |
| NDD |
Neurodevelopmental disorders |
| NF2 |
NF2, moesin-ezrin-radixin like (MERLIN) tumor suppressor |
| NGS |
Next-generation sequencing |
| NOP56 |
NOP56 ribonucleoprotein |
| NOTCH1 |
Notch receptor 1 |
| NOTCH2NLC |
Notch 2 N-terminal like C |
| OB-GYN |
Obstetrician-gynecologist |
| OTC |
Over the counter |
| PABPN1 |
Poly(a) binding protein nuclear 1 |
| PCSK9 |
Proprotein convertase subtilisin/kexin type 9 |
| PDE3A |
Phosphodiesterase 3A |
| pES |
Prenatal exome sequencing |
| PFS |
Progression-free survival |
| PHOX2B |
Paired like homeobox 2B |
| PICU |
Pediatric intensive care unit |
| PKP2 |
Plakophilin 2 |
| PMS2 |
PMS1 homolog 2, mismatch repair system component |
| PNPLA6 |
Patatin like phospholipase domain containing 6 |
| POLR3A |
RNA polymerase III subunit A |
| PPP2R2B |
Protein phosphatase 2 regulatory subunit Beta |
| PQF |
Perinatal Quality Foundation |
| PRKAG2 |
Protein kinase AMP-activated non-catalytic subunit gamma 2 |
| PROKR2 |
Prokineticin receptor 2 |
| PTEN |
Phosphatase and tensin homolog |
| RB1 |
RB transcriptional corepressor 1 |
| RELN |
Reelin |
| RET |
Ret proto-oncogene |
| rWGS |
Rapid whole genome sequencing |
| RYR1 |
Ryanodine receptor 1 |
| RYR2 |
Ryanodine receptor 2 |
| SCN5A |
Sodium voltage-gated channel alpha subunit 5 |
| SDHAF2 |
Succinate dehydrogenase complex assembly factor 2 |
| SDHB |
Succinate dehydrogenase complex iron sulfur subunit B |
| SDHC |
Succinate dehydrogenase complex subunit C |
| SDHD |
Succinate dehydrogenase complex subunit D |
| SFM/SMFM |
Society for Maternal and Fetal Medicine |
| SLIT2 |
Slit guidance ligand 2 |
| SMAD3 |
SMAD family member 3 |
| SMAD4 |
SMAD family member 4 |
| SPRED3 |
Sprouty related EVH1 domain containing 3 |
| STK11 |
Serine/threonine kinase 11 |
| TCF4 |
Transcription factor 4 |
| TGFBR1 |
Transforming growth factor beta receptor 1 |
| TGFBR2 |
Transforming growth factor beta receptor 2 |
| TMEM43 |
Transmembrane protein 43 |
| TNNI3 |
Troponin I3, cardiac type |
| TNNT2 |
Troponin T2, cardiac type |
| TP53 |
Tumor protein p53 gene |
| TPM1 |
Tropomyosin 1 |
| TRAPPC9 |
Trafficking protein particle complex subunit 9 |
| TSC1 |
TSC complex subunit 1 |
| TSC2 |
TSC complex subunit 2 |
| VHL |
von Hippel-Lindau tumor suppressor gene |
| VUS |
Variants of unknown significance |
| WES |
Whole exome sequencing |
| WGS |
Whole genome sequencing |
| WT1 |
WT1 transcription factor |
Rationale
DNA sequencing is a critical tool for the evaluation of many medical conditions. The two primary methods of DNA sequencing in the clinical setting are Sanger sequencing and next-generation sequencing or NGS. NGS is a technique that allows for the rapid sequencing of multiple strands of DNA. It is not limited to one specific type of test; rather it encompasses numerous technologies that produce swift and high-volume sequencing. NGS can be used to sequence larger sequences, such as the exome or the entire genome. This is opposed to the traditional Sanger sequencing, which is more useful for sequencing a specific gene (Hulick, 2024).
The NGS procedure typically includes the following steps: first the patient’s DNA is prepared to serve as a template, then DNA fragments are isolated (on solid surfaces such as small beads) where sequence data is generated. Then these results are compared against a reference genome. Any DNA sample may be used if the quality and quantity of that sample is sufficient, but the methods of library generation and data analysis often vary from panel to panel. Evaluating the results of a gene panel typically requires expertise in bioinformatics. Since NGS reports data on any variants found, great care must be taken to evaluate these gene variants, especially variants of unknown significance (VUS) and secondary findings (Hulick, 2024; Rehm et al., 2013).
Exome and genome sequencing are usually performed with NGS. The exome represents all the protein-encoding genes, and at least 85% of pathogenic mutations are found in the exome. Further, the exome only comprises approximately 1.5% – 2% of the genome, thereby making it more cost effective to sequence than whole genome sequencing. The entire exome includes approximately 30 megabases compared to the genome’s 3.3 gigabases. However, sequencing an entire genome may be useful as a pathogenic mutation may be in a noncoding region of the genome, such as gene regulation dysfunction. Most clinical NGS testing uses targeted panels or whole exome sequencing (WES), and whole genome sequencing (WGS) is only used in select cases. For instance, conditions such as nonsyndromic hearing loss (possible pathogenic variants in over 60 genes) may benefit from WES evaluation (Hulick, 2024).
Several companies have pivoted towards focused exome sequencing. These are panels tailored to individual phenotypes and target a maximum number of genes depending on the company. There is a > 30% diagnostic yield, with freedom for clinicians to choose specific genes they are interested in, with reduced cost and options to reflex to WES for negative cases (GGC, 2022). In their study, Jia et al. (2023) retrospectively analyzed 372 pediatric patients who were referred to clinically focused exome sequencing (CFES), and concluded that CFES may be first-line for “diagnosing young children with suspected genetic conditions, as it validates the identification of molecular genetics alterations and facilitates comprehensive medical management. The patients that were more likely to receive diagnoses via CFES were those with “metabolism/homeostasis abnormalities, craniofacial /otolaryngology/ ophthalmologic abnormalities, and/or [abnormalities of] the integument” (Jia et al., 2023). Despite the novelty and expected benefits of focused exome sequencing, more clinical studies with larger sample sizes are necessary.
Proprietary Testing
Many proprietary technologies for WES and WGS are available. Companies such as Variantyx provide highly specialized genetic testing to patients and clinicians. The Genomic Unity® Exome Plus Analysis test sequences the exome, including intronic and regulatory variants, identifies disease causing deletions or duplications in the genome, and analyzes the mitochondrial genome with heteroplasmy (≥5%). This test may identify many genes for a variety of disorders including AR, ATN1, ATXN1, ATXN2, ATXN3, ATXN7, ATXN8OS, ATXN10, C9ORF72, CACNA1A, CNBP, CSTB, DMPK, FMR1, FXN, HTT, JPH3, NOP56, NOTCH2NLC, PABPN1, PPP2R2B, TBP (for adult-onset movement disorders), AFF2, DIP2B, FMR1 (for early-onset intellectual disability disorders), and PHOX2B, TCF4 (for other disorders) (variantyx, 2020). This test requires either a blood, saliva or gDNA (genomic DNA) sample and has an eight-week turnaround time.
Clinical Utility and Validity
Alfares et al. (2018) compared the cost-effectiveness and clinical utility of both WES and WGS. Data was analyzed from 108 participants; all 108 individuals had negative array comparative genomic hybridization (also known as chromosomal microarray) results and negative or inconclusive WES results before WGS was performed. Chromosomal microarray (CMA) is another common genetic testing method that can analyze many pieces of DNA simultaneously. The authors only pinpointed three positive cases where WGS identified a genetic or inherited disorder that WES did not recognize; further, it was noted that “30% of the positive cases identified by WGS could be identified by reanalyzing the WES raw data, and WGS achieved an only 7% higher detection rate. Therefore, until the cost of WGS approximates that of WES, reanalyzing WES raw data is recommended before performing WGS” (Alfares et al., 2018).
Yang et al. (2014) conducted a single-center observational study of 2000 patients with clinical whole exome sequencing performed for a suspected genetic disorder. A molecular diagnosis was reported for 504 patients (25.2%) with 58% of the diagnostic mutations not previously reported. The investigators concluded that “the yield of whole-exome sequencing may offer advantages over traditional molecular diagnostic approaches in certain patients” (Yang et al., 2014). Best et al. (2018) reviewed 31 different WES studies and noted that the diagnostic rates varied between 6.2% and 80%; however, the researchers state that the “differences in inclusion criteria and trio versus singleton approaches to sequencing largely account for the wide range of diagnostic rates.”
Tammimies et al. (2015) evaluated the molecular diagnostic yield of CMA and WES in children with autism spectrum disorder (ASD). The patient cohort included 258 consecutively enrolled unrelated children with ASD, stratified into three groups based on the presence of major congenital abnormalities and minor physical anomalies. All probands underwent CMA, with WES performed for 95 proband-parent trios. The molecular diagnostic yields of CMA and WES were comparable. Among the 95 patients undergoing WES, eight children (8.4%) received an ASD-related molecular diagnosis. Among the children who underwent both CMA and WES testing, the estimated proportion with an identifiable genetic etiology was 15.8%. The investigators concluded that “if replicated in additional populations, these findings may inform appropriate selection of molecular diagnostic testing for children affected by ASD” (Tammimies et al., 2015). A similar study was performed by Arteche-López et al. (2021) to validate WES as a “first-tier test for the genetic diagnosis of [ASD], when there is no suspicion of fragile X syndrome.” Upon comparing the clinical utility of CMA, FMR1 testing, and WES testing, the researchers “achieved a global diagnostic rate of 12.8% (44/343), the majority of them being characterised by WES (33/44; 75%) compared to CMA (9/44; 20.4%) or FMR1 testing (2/44; 4.5%),” evidently demonstrating the “higher diagnostic power” of WES compared to CMA (Arteche-López et al., 2021).
Taylor et al. (2015) performed whole genome sequencing in 217 individuals across a broad spectrum of genetic disorders in whom previous screening had identified no pathogenic variants. Disease-causing variants were identified in 21% of cases, with the proportion increasing to 34% (23/68) for mendelian disorders and 57% (8/14) in family trios. The investigators concluded that the results “demonstrate the value of genome sequencing for routine clinical diagnosis but also highlight many outstanding challenges” (Taylor et al., 2015).
Miller et al. (2017) performed exome/whole genome sequencing to identify the genetic cause in patients with craniosynostosis, in whom prior clinically driven genetic testing had been negative. Out of the 40 patients’ tests, associated mutations were identified in 15 patients (37.5%) involving 14 different genes. In five of the 15 positive cases, the molecular diagnosis had immediate, actionable consequences in patient management. The investigators concluded that “the benefits of exome/whole genome sequencing to identify causal mutations in craniosynostosis cases for which routine clinical testing has yielded negative results” (Miller et al., 2017).
Crowley et al. (2020) used WES in a single-center cohort study of 1005 pediatric IBD patients and found a 3% prevalence of damaging variants in genes linked to monogenic IBD, and that 1% of monogenic pediatric IBD patients have variants in genes associated with primary immunodeficiency that are potentially curable through allogeneic hematopoietic stem cell transplantation. As rare genetic variants could manifest in different phenotypes, the researchers believe that the “data supports the diagnosis of monogenic disease beyond the very early onset IBD population especially in children with a family history of autoimmune diseases and those with evidence of extra-intestinal manifestations of IBD” and that WES will lend itself to provide definitive and personalized treatments in the future (Crowley et al., 2020).
Srivastava et al. (2014) conducted a retrospective cohort study on 78 children with neurodevelopmental disabilities and a prior unrevealing workup before WES. The overall presumptive diagnostic testing rate was 41% (32/78 patients). Results of WES affected patient management in all cases, most often related to reproductive planning (27/78). The investigators concluded that the high diagnostic yield of WES could lead to earlier diagnosis, impacting medical management, prognostication, and family planning (Srivastava et al., 2014).
Haskell et al. (2018) studied the diagnostic utility of exome sequencing in the evaluation of neuromuscular disorders. A total of 93 undiagnosed patients with potential neuromuscular disorders participated in this study; the diagnostic yield of these 93 patients with exome sequencing was 12.9% “with one or more pathogenic or likely pathogenic variants identified in a causative gene associated with the patient's disorder” (Haskell et al., 2018). In this study, exome sequencing was able to provide or clarify a neuromuscular disorder diagnosis, but only in a small percentage of the population studied.
Involving WGS or WES as a supplemental level of evaluation has been able to effect change in medical care and treatment pathways. In the NIH-funded Undiagnosed Diseases Network, among 382 patients with complete evaluations, “28 (21%) of the patients who received a diagnosis, the diagnosis led to a recommendation regarding a change in therapy. In 49 (37%), the diagnosis led to a change in care other than therapy, such as the narrowing of diagnostic testing. In 48 (36%), the diagnosis led to variant-specific genetic counseling but did not lead to a change in the diagnostic or therapeutic strategy.” The changes in therapy ranged from known drugs, vitamins, coenzyme supplementations, and transplant in one patient. This demonstrated evidence supporting usage of DNA sequencing for genetically determined conditions and a representative lens of how it can affect medical care (Splinter et al., 2018).
Muthaffar (2021) conducted a retrospective chart review for WES results between January 2018 to November 2019 for patients at a pediatric neurology clinic in Saudi Arabia to identify the utility of WES. It was found that “twenty-six children with undiagnosed neurological conditions were identified and underwent WES diagnosis. Nineteen patients (73.0%) of the cohort were diagnosed with pathogenic variants, likely pathogenic variants or variants of unknown significance (VUS).” The researcher also furthered the conclusions of the WES high diagnostic rate by proving direct implications on clinical management based on testing results. One patient who had a positive pathogenic BTD mutation, diagnosed at seven-years old with a biotinidase deficiency, was started on biotin supplements after WES testing, and was able to breathe independently off a ventilator, regain motor capabilities with physical therapy, improve hearing, and eliminate convulsions (Muthaffar, 2021).
Using WES on 11 probands from 10 Jordanian families who have been formerly diagnosed with limb-girdle dystrophy (LGMD) and Charcot-Marie-Tooth disease (CMT), Ababneh et al. (2021) identified a series of missense, nonsense, and deletion variants associated with neuromuscular disorders. Consequently, the researchers argue that “Utilization of WES is helpful to facilitate rapid and accurate NMDs diagnosis, complementing a thorough clinical evaluation”, especially in a country where the risk of autosomal-recessive disorders is increased by consanguinity and the implementation of genetic diagnosis is limited and the results misunderstood (Ababneh et al., 2021).
Sanford et al. (2019) investigated the clinical utility of rWGS in children within the pediatric intensive care unit (PICU). They were able to make a molecular diagnosis by rWGS in 17 of 38 children, and in four of the 17 children diagnosed by rWGS (24%), “the genetic diagnoses led to a change in management while in the PICU, including genome-informed changes in pharmacotherapy and transition to palliative care. … Eighty-two percent of diagnoses affected the clinical management of the patient and/or family after PICU discharge, including avoidance of biopsy, administration of factor replacement, and surveillance for disorder-related sequelae” (Sanford et al., 2019). In this retrospective analysis, benefits of rWGS were further elucidated in the setting of unknown or unclear clinical etiologies.
Reda et al. (2020) studied WES for metastatic solid cancer diagnoses in 506 patients. In this study, the somatic and germline exome analysis was restricted to 317 specific genes. Exome sequencing was successful in 386 tumor samples, and 342 patients received a therapeutic proposal based on their genetic results. However, only 79 patients were treated with an NGS matched therapy. While this study shows that WES is a possible tool to assist with metastatic solid cancer diagnoses and treatments, “no differences were observed between PFS [progression-free survival] ratios of patients treated with matched therapy versus standard therapy” (Reda et al., 2020).
Other studies have also yielded bifurcating results on the periodic revisiting of unsolved exome cases and for variants of unknown significance. Salfati et al. (2019) found that re-analysis of 101 WES cases one to seven years after initial analysis resulted in “the identification of additional diagnostic variants in 3 rare disease cases (5.9%) and 1 sudden unexplained death case (2%), which increased our molecular diagnostic yield to 31.4% and 12%, respectively.” However, though they recognize the importance of any diagnostic yield to those families potentially affected, the authors also acknowledge that “most of our cases remain unexplained after our re-analysis,” which they attribute to an enduring lack of coverage of functional exonic variants, along with “the possibility of complicated oligogenic disease that is not easily dissected in small families, and the possibility of disease due to epigenetic, somatic, or other uninterrogated genomic aberrations.” As such, “We [the authors] suggest that a 6-month cycle of automated re-analysis could improve the pace at which new findings are disseminated to patients. Periodic re-analysis by third party or other software not originally used to analyze cases is also potentially useful to uncover pathogenic variants that may be missed by the differences across genome interpretation platforms” (Salfati et al., 2019).
Parent-child Trio Testing
Parent-child trio testing is a strategy which helps to identify single pathogenic mutations among the many genomic variants in an individual. Specifically, the sequencing of both the parents and the patient allows for the variant to be identified easier and “filtered based on consistency or inconsistency according to the laws of Mendelian inheritance” (Sakai et al., 2013).
Lee et al. (2014) reported on the initial clinical indications for clinical exome sequencing (CES) referrals and molecular diagnostic rates for different indications and different test types. CES was performed on 814 patients with undiagnosed, suspected genetic conditions who underwent WES. CES was conducted using a trio-CES technique which involves both parents and their affected child sequenced simultaneously. Overall, a molecular diagnosis with a causative variant in a well-established clinical gene was provided for 213/814 (26%) cases. The trio-CES was associated with a higher molecular diagnostic yield (31%; 127/410 cases) than proband-CES or traditional molecular diagnostic methods. The investigators concluded that “additional studies designed to validate these findings and to explore the effect of this approach on clinical and economic outcomes are warranted” (Lee et al., 2014).
Soden et al. (2014) performed diagnostic WGS and/or WES in parent-child trios for 100 families with 119 children with neurodevelopmental disorders (NDD). A total of 45% of the families received molecular diagnoses of an established genetic disorder (53/119 affected children). An accelerated sequencing modality, rapid WGS, yielded diagnoses in 73% of families with acutely ill children (11/15). In this study, WES proved to be less costly than continued conventional diagnostic testing of children with NDD in whom initial testing failed to yield a diagnosis. The investigators concluded that “initial diagnostic evaluation of children with NDD should include trio WGS or WES, with extension of accelerated sequencing modalities to high-acuity patients” (Soden et al., 2014).
Another study compared fetal WES versus trio analysis WES on fetuses with sonographic abnormalities. The researchers found that trio analysis yielded a positive/definitive diagnosis in 30% (3/10) of the cases as compared to only 14.3% (2/14) of the singleton cases. They conclude, “In order to expedite interpretation of results, trio sequencing should be employed, but interpretation can still be compromised by incomplete coverage of relevant genes” (Drury et al., 2015). Similarly, these data are supported by another study of trio analysis of thirty different cases. A total of 10% of the cases were positive for a pathogenic finding, and 17% were de novo, inherited recessive, or X-linked variants. The authors conclude, “This study outlines the way for a substantial improvement in the diagnostic yield of prenatal genetic abnormalities through the application of next-generation sequencing” (Carss et al., 2014).
Yates et al. (2017) performed WES, including trio analysis, using samples obtained from deceased fetuses with ultrasound anomalies. They note that 20% of cases were positive overall with a definitive diagnosis with another 45% positive for possible pathogenic candidate variants. Comparing trio analysis to singleton analysis, 24% (n = 11) of trio analysis resulted in a definitive diagnostic finding versus 14% (n = 3) for singleton testing (Yates et al., 2017).
Clark et al. (2018) compared the diagnostic and clinical utility of WGS, WES and CMA in children with suspected genetic disorders. Trio analyses were also analyzed. Many studies were reviewed in this meta-analysis; the authors state that “In 37 studies, comprising 20,068 children, diagnostic utility of WGS (0.41, 95% CI 0.34 – 0.48, I2 = 44%) and WES (0.36, 95% CI 0.33 – 0.40, I2 = 83%) were qualitatively greater than CMA (0.10, 95% CI 0.08 – 0.12, I2 = 81%) (Clark et al., 2018). Further, a statistical difference was not found regarding the diagnostic utility of WES and WGS. Finally, “Subgroups with higher WGS/WES diagnostic utility were trios and those receiving hospital-based interpretation. WGS/WES should be considered a first-line genomic test for children with suspected genetic diseases” (Clark et al., 2018).
Zhang et al. (2021) performed WES and trio analysis on 18 unrelated men who have idiopathic hypogonadotropic hypogonadism (IHH), which is a rare genetic disorder that causes delayed or absent puberty as well as infertility due to gonadotropin-releasing hormone (GnRH) insufficiency/deficiency, and their parents. With this testing, “one reported and 10 novel variants in eight known IHH causative genes (AXL, CCDC141, CHD7, DMXL2, FGFR1, PNPLA6, POLR3A, and PROKR2), nine variants in nine recently reported candidate genes (DCAF17, DCC, EGF, IGSF10, NOTCH1, PDE3A, RELN, SLIT2, and TRAPPC9), and four variants in four novel candidate genes for IHH (CCDC88C, CDON, GADL1, and SPRED3) were identified in 77.8% (14/18) of IHH cases.” This analysis also supported oligogenic etiology for disease presentation, with 44.4% cases carrying at least two variants in IHH-related genes. They also found that the variants “tended to be maternally inherited (maternal with n = 17 vs paternal with n = 7; P = 0.028),” which was confirmed by their previous literature review, and due to the presence of female carriers, extends the notion that females may be more tolerant of “deleterious” IHH-related gene mutations. This study exemplifies the clinical utility of WES and trio analysis for reproductive genetic disorders and could be used to continue pedigree analyses for IHH (Zhang et al., 2021).
Malcher et al. (2022) investigated the use of whole-genome sequencing in identifying new candidate genes for nonobstructive azoospermia (NOA). The authors applied WGS for 39 patients with NOA to identify novel NOA-associated SNVs, yielding “8 potentially disease causing [sic] variants in 4 genes, followed by 30 variants in 20 genes that were previously linked to infertility, and 20 variants in 13 genes that have never been investigated with respect to male infertility but could be important in patients with NOA” in 29 of the 39 azoospermic individuals. Of these 58 variants, 16 were newly discovered and, as such, “highly recommended to examine their possible function and mechanism of participation in gametogenesis” (Malcher et al., 2022).
In their examination of whole exome and genome sequencing in a Mendelian disorder cohort, Ewans et al. (2022) determined that “WGS resulted in a diagnosis in one third (34%; 13/38 families) of undiagnosed families who had previously had WES.” However, when adjusting for “factors such as improvements to gene-disease knowledge and genomic pipelines through contemporary WES reanalysis, the WGS diagnostic yield reduced to 19% (6/31 remaining families)”, primarily due to “due to reduced WES coverage of critical regions that may be solved through an improved WES platform.” Such results contribute to the debate about the “trade-off between the lower cost of WES and the higher diagnostic yield of WGS” and will ultimately be a function of “the clinical scenario and local resourcing and availability” (Ewans et al., 2022).
American College of Medical Genetics (ACMG)
In 2012, the ACMG released a policy statement outlining points to consider in the clinical application of genomic sequencing to the detection of germ-line mutations. The ACMG recommended that WGS/WES should be considered in the clinical diagnostic assessment of a phenotypically affected individual when:
- “The phenotype or family history data strongly implicate a genetic etiology, but the phenotype does not correspond with a specific disorder for which a genetic test targeting a specific gene is available on a clinical basis.”
- “A patient presents with a defined genetic disorder that demonstrates a high degree of genetic heterogeneity, making WES or WGS analysis of multiple genes simultaneously a more practical approach.”
- “A patient presents with a likely genetic disorder, but specific genetic tests available for that phenotype have failed to arrive at a diagnosis.”
- “A fetus with a likely genetic disorder in which specific genetic tests, including targeted sequencing tests, available for that phenotype have failed to arrive at a diagnosis.”
ACMG stated that “WGS/WES may be considered in preconception carrier screening, using a strategy to focus on genetic variants known to be associated with significant phenotypes in homozygous or hemizygous progeny.” ACMG further stated that WGS and WES should not be used at this time as an approach to prenatal screening or as a first-tier approach for newborn screening (ACMG, 2012).
ACMG released a guideline on informed consent for genome/exome sequencing. In that guideline, they noted that WGS/WES was not recommended “before the legal age of majority” unless for “phenotype-driven clinical diagnostic uses or circumstances in which early monitoring or interventions are available and effective” (ACMG, 2013).
In 2014 the ACMG published guidelines (Alford et al., 2014) for the clinical evaluation and etiologic diagnosis of hearing loss which state: “Pretest genetic counseling should be provided, and, with patient's informed consent, genetic testing, if available, should be ordered to confirm the diagnosis — this testing may include single-gene tests, hearing loss sequencing panels, whole-exome sequencing(WES), whole-genome sequencing (WGS), chromosome analysis, or microarray-based copy-number analysis, depending on clinical findings.”
In 2020 the ACMG published guidelines on the use of fetal exome sequencing (ES) in prenatal diagnoses. These guidelines are below (Monaghan et al., 2020):
Pretest Considerations
- “Exome sequencing may be considered for a fetus with ultrasound anomalies when standard CMA and karyotype analysis have failed to yield a definitive diagnosis. If a specific diagnosis is suspected, molecular testing for the suggested disorder (with single-gene test or gene panel) should be the initial test. At the present time, there are no data supporting the clinical use for ES for other reproductive indications, such as the identification of sonographic markers suggestive of aneuploidy or a history of recurrent unexplained pregnancy loss.
- Trio analysis consisting of the proband and both biological parents is preferred to singleton (fetus only) or duo (fetus and one parent) analyses. Trio analysis consistently shows higher diagnostic yields compared with nontrio analysis. It allows for the immediate identification of de novo variants, determination of phase for biallelic variants, and confirmation of carrier status in both parents when a homozygous variant is detected. For laboratories not requiring trio analysis for prenatal ES, all efforts should be made to determine inheritance of identified fetal variants with targeted testing of the biological parents. There may be circumstances where both biological parents are unable to submit specimens. In this scenario, variant segregation testing using the available parent or testing of other closely related family members should be considered.
- Pretest counseling is ideally provided by a genetics professional during which the types of variants that may be returned in a laboratory report for all tested family members would be reviewed.
Posttest Considerations
- Post-test counseling is recommended, regardless of the test result. It should be provided by individuals with relevant expertise, preferably a genetics professional.
Reanalysis Considerations
- For patients with initial negative ES results, reanalysis of exome sequencing data aids clinical diagnosis after 12 months. This outcome has been validated in the pediatric population as additional phenotypic findings might be noted during a child’s growth and development. Continuous updates in database resources and new publications may provide further information for variant and gene classification.
- Due to the discovery of new gene–disease associations (that were unknown at the time of initial analysis), reanalysis can also be considered for diagnostic results and results deemed to be possibly (but not definitively) associated with the fetal phenotype.
- For fetal ES with nondiagnostic or negative results, reanalysis may be considered if a new phenotype develops in the proband after birth, a future pregnancy is planned, or a significant amount of time has passed (either at the discretion of the testing laboratory or at least 12 months) since the initial testing was performed.
- If the original prenatal ES report does not account for the complete phenotype or if new/additional phenotypes develop over time, a reanalysis could be considered” (Monaghan et al., 2020).
In 2020 the ACMG conducted a systematic evidence review to support guideline development for the use of exome and genome sequencing among patients with congenital anomalies, developmental delay, or intellectual disability (CA/DD/ID). From their review, the ACMG concluded, “There is evidence that ES/GS for patients with CA/ DD/ID informs clinical and reproductive decision-making, which could lead to improved outcomes for patients and their family members. Further research is needed to generate evidence regarding health outcomes to inform robust guidelines regarding ES/GS in the care of patients with CA/DD/ID” (Malinowski et al., 2020).
In 2021 the ACMG asserted that as the body of literature surrounding this continues to burgeon, it urges them to “strongly recommend ES and GS as a first-tier or second-tier test (guided by clinical judgment and often clinician-patient/family shared decision making after CMA or focused testing) for patients with one or more CAs prior to one year of age or for patients with DD/ID with onset prior to 18 years of age” (Manickam et al., 2021).
American College of Obstetricians and Gynecologists (ACOG) and Society for Maternal and Fetal Medicine (SFM)
In 2016 the ACOG and SFM published a joint committee opinion on “Microarrays and Next-Generation Sequencing Technology: The Use of Advanced Genetic Diagnostic Tools in Obstetrics and Gynecology”, which states that “the routine use of whole-genome or whole-exome sequencing for prenatal diagnosis is not recommended outside of the context of clinical trials until sufficient peer reviewed data and validation studies are published” (ACOG & SFM, 2016).
However, ACOG and SFM note that WES may be considered when “specific genetic tests available for a phenotype, including targeted sequencing tests, have failed to determine a diagnosis in a fetus with multiple congenital anomalies suggestive of a genetic disorder.” The guideline further clarifies that “in select circumstances (recurrent or lethal fetal anomalies in which other approaches have been noninformative), [WES] may be considered as a diagnostic tool, but only after other appropriate testing has been noninformative and after extensive counseling by an [OB-GYN[ or other health care provider with genetics expertise who is familiar with these new technologies and their limitations” (ACOG & SFM, 2016). This committee opinion was reaffirmed in 2023.
Joint Position Statement From the International Society for Prenatal Diagnosis (ISPD), the Society for Maternal Fetal Medicine (SMFM), and the Perinatal Quality Foundation (PQF)
Per the guideline, the word “sequencing” is used to refer to “whole exome sequencing, targeted analysis using clinical panels, and whole genome sequencing.”
“The use of diagnostic sequencing is currently being introduced for evaluation of fetuses for whom standard diagnostic genetic testing, such as chromosomal microarray analysis (CMA), has already been performed and is uninformative or is offered concurrently according to accepted practice guidelines, or for whom expert genetic opinion determines that standard genetic testing is less optimal than sequencing for the presenting fetal phenotype” (ISPD et al., 2018).
Routine use of prenatal sequencing as a diagnostic test cannot be supported due to “insufficient” validation and data about benefits and pitfalls (ISPD et al., 2018).
Within the section on recommendations for all diagnostic applications of genome-wide sequencing, concerning trio analysis, they state, “Diagnostic sequencing for fetal indications is best done as a trio analysis, where fetal and both parental samples are sequenced and analyzed together. The trio approach currently benefits timeliness of result interpretation and aids assignment of pathogenicity for detected sequence variants. If proband‐only sequencing is performed, validation of diagnostic or potentially diagnostic findings best includes a determination of inheritance through targeted testing of samples from biological parents” (ISPD et al., 2018). However, the guideline could not recommend one sequencing method over another, nor was the guideline certain on the best way to interpret variants found in genome-wide sequencing.
The guideline provides three scenarios in which fetal sequencing may be “beneficial”:
“A current pregnancy with a fetus with a single major anomaly or with multiple organ system anomalies that are suggestive of a possible genetic etiology, but no genetic diagnosis was found after CMA; or in select situations with no CMA result, following a multidisciplinary review and consensus, in which there is a fetus with a multiple anomaly ‘pattern’ that strongly suggests a single gene disorder.”
“A personal (maternal or paternal) history of a prior undiagnosed fetus (or child) affected with a major single anomaly or multiple anomalies suggestive of a genetic etiology, and a recurrence of similar anomalies in the current pregnancy without a genetic diagnosis after karyotype or CMA. In addition, when such parents present for preconception counseling and no sample is available from the affected proband, or if a fetal sample cannot be obtained in an ongoing pregnancy, it is considered appropriate to offer sequencing for both biological parents to look for shared carrier status for autosomal recessive mutations that might explain the fetal phenotype. However, where possible, obtaining tissue from a previous abnormal fetus or child for exome sequencing is preferable.”
“In families with a history of recurrent stillbirths of unknown etiology after karyotype and/or CMA, where the fetus in the current pregnancy has a recurrent pattern of anomalies (ISPD et al., 2018).
International Society for Prenatal Diagnosis (ISPD)
In 2022, the ISPD released an updated position statement on the use of genome-wide sequencing for prenatal diagnosis. Below are the pertinent recommendations:
- “Diagnostic sequencing for fetal indications is best done as a trio analysis, where fetal and both parental samples are sequenced and analyzed together.”
- “Approaches to sequence analysis may vary from examination of genes known to be associated with fetal or neonatal phenotypes to a broader genome-wide strategy. It is also uncertain whether interpretation of variants found by genome-wide sequencing should follow the general guidelines for interpretation and reporting of results for children and adults, or whether a more restrictive approach, limited to those variants that explain the phenotype is preferable in the prenatal setting, or if a new approach restricting reporting to severe childhood conditions should be considered.”
- “The current existing data support that prenatal sequencing is beneficial for the following indications:
- A current pregnancy with a fetus having a major single anomaly or multiple organ anomalies:
- For which no genetic diagnosis was found after CMA and a clinical genetic expert review considers the phenotype suggestive of a possible genetic etiology.
- For which the multiple anomaly “pattern” strongly suggests a single gene disorder with no prior genetic testing. As pES is not currently validated to detect all CNVs, CMA should be run before or in parallel with pES in this scenario.
- A personal (maternal or paternal) history of a prior undiagnosed fetus (or child) affected with a major single or multiple anomalies:
- With a recurrence of similar anomalies in the current pregnancy without a genetic diagnosis after karyotype or CMA for the current or prior undiagnosed pregnancy.
- When such parents present for preconception counseling and no sample is available from the affected proband, or if a fetal sample cannot be obtained in an ongoing pregnancy, it is considered appropriate to offer sequencing for both biological parents to look for shared carrier status for autosomal recessive mutations that might explain the fetal phenotype. However, where possible, obtaining tissue from a previous abnormal fetus or child for pES is preferable.
- A current pregnancy with a fetus having a major single anomaly or multiple organ anomalies:
- There is currently no evidence that supports routine testing (including upon parental request) on fetal tissue obtained from an invasive prenatal procedure (amniocentesis, CVS, cordocentesis, other) for indications other than fetal anomalies
- There may be special settings when prenatal sequencing in the absence of a fetal phenotype visible on prenatal imaging can be considered, such as with a strong family history of a recurrent childhood‐onset severe genetic condition with no prenatal phenotype in previous children for whom no genetic evaluation was done and is not possible. Such scenarios should be reviewed by an expert multidisciplinary team preferentially in the context of a research protocol. If sequencing is done for this indication, it must be done as trio sequencing, using an appropriate analytical approach” (Van den Veyver et al., 2022).
American Academy of Neurology (AAN) and American Association of Neuromuscular and Electrodiagnostic Medicine (AANEM)
The AAN/AANEM published guidelines (Kang et al., 2015) on the evaluation, diagnosis, and management of congenital muscular dystrophy (CMD) which state: “In individuals with CMD who either do not have a mutation identified in one of the commonly associated genes or have a phenotype whose genetic origins have not been well characterized, physicians might order whole-exome or whole-genome sequencing when those technologies become more accessible and affordable for routine clinical use (Level C).”
The AAN/AANEM published guidelines (Narayanaswami et al., 2014) on the diagnosis and treatment of limb-girdle and distal dystrophies which state: “In patients with suspected muscular dystrophy in whom initial clinically directed genetic testing does not provide a diagnosis, clinicians may obtain genetic consultation or perform parallel sequencing of targeted exomes, whole-exome sequencing, whole-genome screening, or next-generation sequencing to identify the genetic abnormality (Level C).”
Association for Molecular Pathology (AMP)
The AMP published a report on the spectrum of clinical utilities in molecular pathology testing procedures for inherited conditions and cancer. The background of this report states, “Whole genome sequencing is currently more expensive than WES, requires greater analysis, and generates more variants of uncertain significance. WES is a plausible approach when the clinical picture cannot be affirmed using a specific gene panel. As technologies and understanding of variants advance, whole genome sequencing might become the test of choice” (Joseph et al., 2016).
American Academy of Pediatrics (AAP)
AAP published guidelines on the evaluation of children with autism spectrum disorder. According to the guidelines, CMA is recommended if the etiology for developmental disability is not known. Since Fragile X Syndrome increases the risk for autism spectrum disorder, DNA testing for Fragile X should be recommended in all children with ASD, especially for boys and children with a family history of intellectual disability. “The cytosine-guanine-guanine trinucleotide repeat expansion that is responsible for fragile X syndrome is not detected on CMA and must be ordered as a separate test. When the history and physical examination, CMA, and fragile X analysis do not identify an etiology, the next step at this time in the etiologic evaluation for [autism spectrum disorder] is whole-exome sequencing (WES).” AAP does not recommend the use of commercially marketed tests as they do not provide a molecular etiologic diagnosis (Hyman et al., 2020).
Canadian College of Medical Geneticists
In 2015, the Canadian College of Medical Geneticists published a position statement on genome-wide sequencing for monogenic diseases. Their relevant recommendations include the following:
- “Recommendations for diagnostic assessment:
- Clinical exome sequencing, at this time, should only be used to interrogate the genome for nucleotide sequence variants in genes known to cause disease. Clinical WGS may be used to detect CNV and structural variation in addition to sequence variants, though it is not currently a first-tier test for such analyses.
- Clinical genome-wide sequencing should be considered in the investigation of an affected individual when his/her phenotype or family history suggests a monogenic aetiology in whom the causal mutation(s) are unknown, and one or more of the following additional conditions apply:
- the phenotype is associated with a high degree of genetic heterogeneity;
- specific genetic tests have failed to arrive at a diagnosis and testing of other clinically relevant genes is appropriate;
- genome-wide sequencing is a more cost-effective approach than available individual gene or gene panel testing.”
- “Until the benefits of reporting incidental findings are established, we do not endorse the intentional clinical analysis of disease-associated genes other than those linked to the primary indication.”
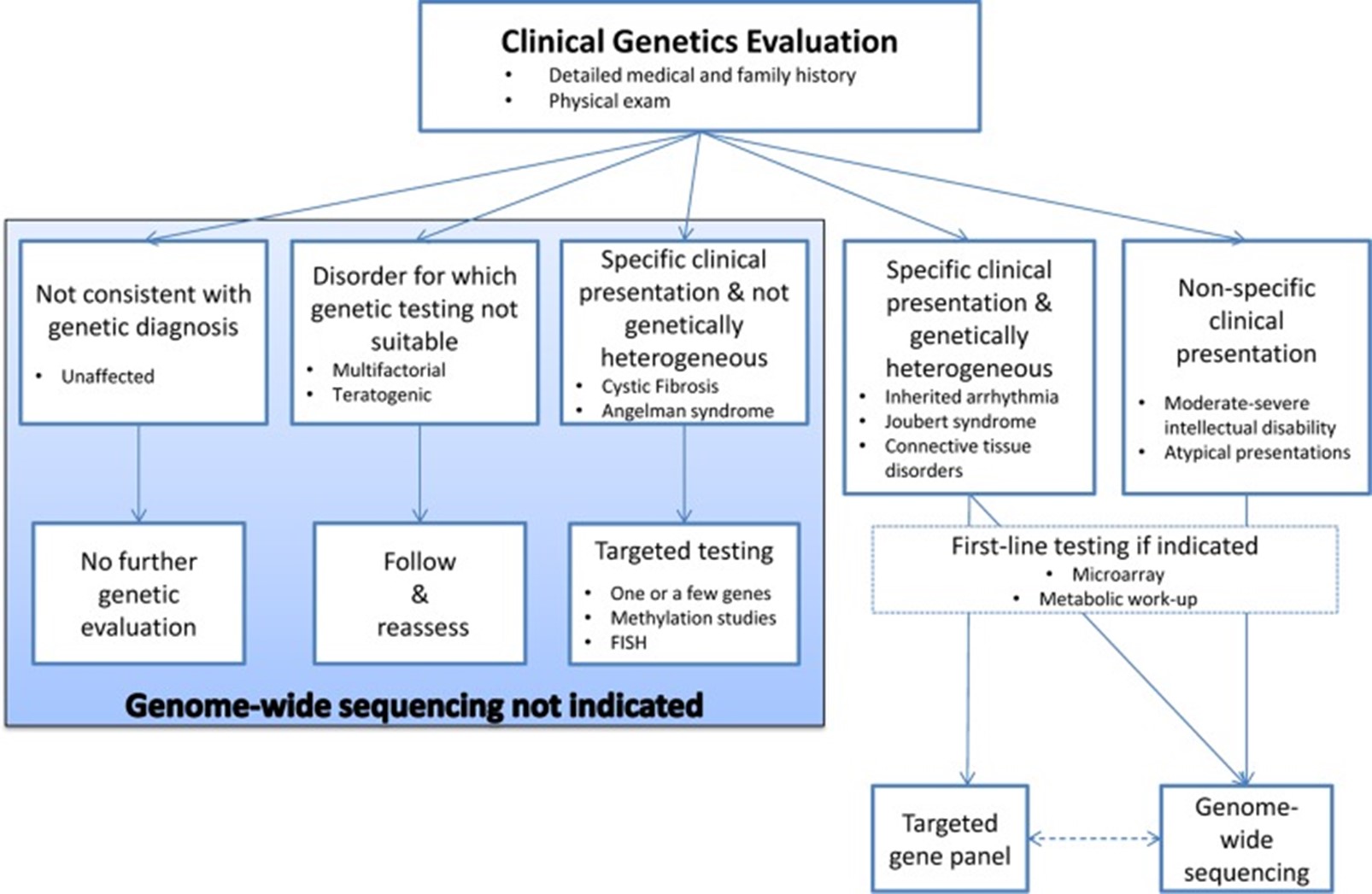
Below is a figure of a “decision aid to facilitate the diagnostic evaluation of patients with rare disease of suspected monogenic aetiology” (Boycott et al., 2015).
The Royal Australasian College of Physicians
In 2021, the Royal Austalasian College of Physicians provided guidelines on pediatric genetic testing in the context of intellectual disability (ID) and global developmental delay (GDD). For childhood syndromes or ID/GDD, the group recommends WES or WGS as tests of choice, since they offer “a broad, agnostic screen,” but acknowledge that WES is more widely available and cost-effective at the time of publication. In terms of ordering singleton or trio testing, the group states that “the latter (trio) approach is highly recommended given it simplifies analysis… it is also a more streamlined clinical test as trio testing identifies fewer variants of uncertain significance than singleton testing” (Sachdev et al., 2021).
References
- Ababneh, N. A., Ali, D., Al-Kurdi, B., Barham, R., Bsisu, I. K., Dababseh, D., Arafat, S., Khanfar, A. N., Makahleh, L., Ryalat, A. T., Sallam, M., El-Khateeb, M., Sharrack, B., & Awidi, A. (2021). The utility of whole-exome sequencing in accurate diagnosis of neuromuscular disorders in consanguineous families in Jordan. Clin Chim Acta, 523, 330-338. https://doi.org/10.1016/j.cca.2021.10.001
- ACMG. (2012). Points to consider in the clinical application of genomic sequencing. Genet Med, 14(8), 759-761. https://doi.org/10.1038/gim.2012.74
- ACMG. (2013). Points to consider for informed consent for genome/exome sequencing [ACMG Policy Statement]. Genetics In Medicine, 15, 748. https://doi.org/10.1038/gim.2013.94
- ACOG, & SFM. (2016). Committee Opinion No.682: Microarrays and Next-Generation Sequencing Technology: The Use of Advanced Genetic Diagnostic Tools in Obstetrics and Gynecology. Obstet Gynecol, 128(6), e262-e268. https://doi.org/10.1097/AOG.0000000000001817
- Alfares, A., Aloraini, T., Subaie, L. A., Alissa, A., Qudsi, A. A., Alahmad, A., Mutairi, F. A., Alswaid, A., Alothaim, A., Eyaid, W., Albalwi, M., Alturki, S., & Alfadhel, M. (2018). Whole-genome sequencing offers additional but limited clinical utility compared with reanalysis of whole-exome sequencing. Genet Med, 20(11), 1328-1333. https://doi.org/10.1038/gim.2018.41
- Alford, R. L., Arnos, K. S., Fox, M., Lin, J. W., Palmer, C. G., Pandya, A., Rehm, H. L., Robin, N. H., Scott, D. A., & Yoshinaga-Itano, C. (2014). American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet Med, 16(4), 347-355. https://doi.org/10.1038/gim.2014.2
- Arteche-López, A., Gómez Rodríguez, M. J., Sánchez Calvin, M. T., Quesada-Espinosa, J. F., Lezana Rosales, J. M., Palma Milla, C., Gómez-Manjón, I., Hidalgo Mayoral, I., Pérez de la Fuente, R., Díaz de Bustamante, A., Darnaude, M. T., Gil-Fournier, B., Ramiro León, S., Ramos Gómez, P., Sierra Tomillo, O., Juárez Rufián, A., Arranz Cano, M. I., Villares Alonso, R., Morales-Pérez, P., . . . Alvarez-Mora, M. I. (2021). Towards a Change in the Diagnostic Algorithm of Autism Spectrum Disorders: Evidence Supporting Whole Exome Sequencing as a First-Tier Test. Genes (Basel), 12(4). https://doi.org/10.3390/genes12040560
- Best, S., Wou, K., Vora, N., Van der Veyver, I. B., Wapner, R., & Chitty, L. S. (2018). Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat Diagn, 38(1), 10-19. https://doi.org/10.1002/pd.5102
- Boycott, K., Hartley, T., Adam, S., Bernier, F., Chong, K., Fernandez, B. A., Friedman, J. M., Geraghty, M. T., Hume, S., Knoppers, B. M., Laberge, A. M., Majewski, J., Mendoza-Londono, R., Meyn, M. S., Michaud, J. L., Nelson, T. N., Richer, J., Sadikovic, B., Skidmore, D. L., . . . Armour, C. M. (2015). The clinical application of genome-wide sequencing for monogenic diseases in Canada: Position Statement of the Canadian College of Medical Geneticists. J Med Genet, 52(7), 431-437. https://doi.org/10.1136/jmedgenet-2015-103144
- Carss, K. J., Hillman, S. C., Parthiban, V., McMullan, D. J., Maher, E. R., Kilby, M. D., & Hurles, M. E. (2014). Exome sequencing improves genetic diagnosis of structural fetal abnormalities revealed by ultrasound. Human molecular genetics, 23(12), 3269-3277. https://doi.org/10.1093/hmg/ddu038
- Clark, M. M., Stark, Z., Farnaes, L., Tan, T. Y., White, S. M., Dimmock, D., & Kingsmore, S. F. (2018). Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom Med, 3, 16. https://doi.org/10.1038/s41525-018-0053-8
- Crowley, E., Warner, N., Pan, J., Khalouei, S., Elkadri, A., Fiedler, K., Foong, J., Turinsky, A. L., Bronte-Tinkew, D., Zhang, S., Hu, J., Tian, D., Li, D., Horowitz, J., Siddiqui, I., Upton, J., Roifman, C. M., Church, P. C., Wall, D. A., . . . Muise, A. M. (2020). Prevalence and Clinical Features of Inflammatory Bowel Diseases Associated With Monogenic Variants, Identified by Whole-Exome Sequencing in 1000 Children at a Single Center. Gastroenterology, 158(8), 2208-2220. https://doi.org/10.1053/j.gastro.2020.02.023
- Drury, S., Williams, H., Trump, N., Boustred, C., Lench, N., Scott, R. H., & Chitty, L. S. (2015). Exome sequencing for prenatal diagnosis of fetuses with sonographic abnormalities. Prenat Diagn, 35(10), 1010-1017. https://doi.org/10.1002/pd.4675
- Ewans, L. J., Minoche, A. E., Schofield, D., Shrestha, R., Puttick, C., Zhu, Y., Drew, A., Gayevskiy, V., Elakis, G., Walsh, C., Adès, L. C., Colley, A., Ellaway, C., Evans, C. A., Freckmann, M. L., Goodwin, L., Hackett, A., Kamien, B., Kirk, E. P., . . . Roscioli, T. (2022). Whole exome and genome sequencing in mendelian disorders: a diagnostic and health economic analysis. Eur J Hum Genet, 30(10), 1121-1131. https://doi.org/10.1038/s41431-022-01162-2
- GGC. (2022). Focused NGS - Panel. https://ggc.org/test-finder-item/focused-ngs-panel
- Haskell, G. T., Adams, M. C., Fan, Z., Amin, K., Guzman Badillo, R. J., Zhou, L., Bizon, C., Chahin, N., Greenwood, R. S., Milko, L. V., Shiloh-Malawsky, Y., Crooks, K. R., Strande, N., Tennison, M., Tilley, C. R., Brandt, A., Wilhelmsen, K. C., Weck, K., Evans, J. P., & Berg, J. S. (2018). Diagnostic utility of exome sequencing in the evaluation of neuromuscular disorders. Neurol Genet, 4(1), e212. https://doi.org/10.1212/nxg.0000000000000212
- Hulick, P. (2024, February 7). Next-generation DNA sequencing (NGS): Principles and clinical applications. https://www.uptodate.com/contents/next-generation-dna-sequencing-ngs-principles-and-clinical-applications
- Hyman, S. L., Levy, S. E., & Myers, S. M. (2020). Identification, Evaluation, and Management of Children With Autism Spectrum Disorder. Pediatrics, 145(1). https://doi.org/10.1542/peds.2019-3447
- ISPD, SMFM, & PQF. (2018). Joint Position Statement from the International Society for Prenatal Diagnosis (ISPD), the Society for Maternal Fetal Medicine (SMFM), and the Perinatal Quality Foundation (PQF) on the use of genome-wide sequencing for fetal diagnosis. Prenat Diagn, 38(1), 6-9. https://doi.org/10.1002/pd.5195
- Jia, A., Lei, Y., Liu, D. P., Pan, L., Guan, H. Z., & Yang, B. (2023). A Retrospective Analysis of Clinically Focused Exome Sequencing Results of 372 Infants with Suspected Monogenic Disorders in China. Pharmgenomics Pers Med, 16, 81-97. https://doi.org/10.2147/pgpm.S387767
- Joseph, L., Cankovic, M., Caughron, S., Chandra, P., Emmadi, R., Hagenkord, J., Hallam, S., Jewell, K. E., Klein, R. D., Pratt, V. M., Rothberg, P. G., Temple-Smolkin, R. L., & Lyon, E. (2016). The Spectrum of Clinical Utilities in Molecular Pathology Testing Procedures for Inherited Conditions and Cancer: A Report of the Association for Molecular Pathology. J Mol Diagn, 18(5), 605-619. https://doi.org/10.1016/j.jmoldx.2016.05.007
- Kang, P. B., Morrison, L., Iannaccone, S. T., Graham, R. J., Bonnemann, C. G., Rutkowski, A., Hornyak, J., Wang, C. H., North, K., Oskoui, M., Getchius, T. S., Cox, J. A., Hagen, E. E., Gronseth, G., & Griggs, R. C. (2015). Evidence-based guideline summary: evaluation, diagnosis, and management of congenital muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology, 84(13), 1369-1378. https://doi.org/10.1212/wnl.0000000000001416
- Lee, H., Deignan, J. L., Dorrani, N., Strom, S. P., Kantarci, S., Quintero-Rivera, F., Das, K., Toy, T., Harry, B., Yourshaw, M., Fox, M., Fogel, B. L., Martinez-Agosto, J. A., Wong, D. A., Chang, V. Y., Shieh, P. B., Palmer, C. G., Dipple, K. M., Grody, W. W., . . . Nelson, S. F. (2014). Clinical exome sequencing for genetic identification of rare Mendelian disorders. Jama, 312(18), 1880-1887. https://doi.org/10.1001/jama.2014.14604
- Malcher, A., Stokowy, T., Berman, A., Olszewska, M., Jedrzejczak, P., Sielski, D., Nowakowski, A., Rozwadowska, N., Yatsenko, A. N., & Kurpisz, M. K. (2022). Whole-genome sequencing identifies new candidate genes for nonobstructive azoospermia. Andrology, 10(8), 1605-1624. https://doi.org/10.1111/andr.13269
- Malinowski, J., Miller, D. T., Demmer, L., Gannon, J., Pereira, E. M., Schroeder, M. C., Scheuner, M. T., Tsai, A. C., Hickey, S. E., & Shen, J. (2020). Systematic evidence-based review: outcomes from exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability. Genet Med, 22(6), 986-1004. https://doi.org/10.1038/s41436-020-0771-z
- Manickam, K., McClain, M. R., Demmer, L. A., Biswas, S., Kearney, H. M., Malinowski, J., Massingham, L. J., Miller, D., Yu, T. W., Hisama, F. M., & Directors, A. B. o. (2021). Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genetics In Medicine, 23(11), 2029-2037. https://doi.org/10.1038/s41436-021-01242-6
- Miller, K. A., Twigg, S. R., McGowan, S. J., Phipps, J. M., Fenwick, A. L., Johnson, D., Wall, S. A., Noons, P., Rees, K. E., Tidey, E. A., Craft, J., Taylor, J., Taylor, J. C., Goos, J. A., Swagemakers, S. M., Mathijssen, I. M., van der Spek, P. J., Lord, H., Lester, T., . . . Wilkie, A. O. (2017). Diagnostic value of exome and whole genome sequencing in craniosynostosis. J Med Genet, 54(4), 260-268. https://doi.org/10.1136/jmedgenet-2016-104215
- Monaghan, K. G., Leach, N. T., Pekarek, D., Prasad, P., & Rose, N. C. (2020). The use of fetal exome sequencing in prenatal diagnosis: a points to consider document of the American College of Medical Genetics and Genomics (ACMG). Genet Med. https://doi.org/10.1038/s41436-019-0731-7
- Muthaffar, O. Y. (2021). The Utility of Whole Exome Sequencing in Diagnosing Pediatric Neurological Disorders. Balkan journal of medical genetics : BJMG, 23(2), 17-24. https://doi.org/10.2478/bjmg-2020-0028
- Narayanaswami, P., Weiss, M., Selcen, D., David, W., Raynor, E., Carter, G., Wicklund, M., Barohn, R. J., Ensrud, E., Griggs, R. C., Gronseth, G., & Amato, A. A. (2014). Evidence-based guideline summary: diagnosis and treatment of limb-girdle and distal dystrophies: report of the guideline development subcommittee of the American Academy of Neurology and the practice issues review panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology, 83(16), 1453-1463. https://doi.org/10.1212/wnl.0000000000000892
- Reda, M., Richard, C., Bertaut, A., Niogret, J., Collot, T., Fumet, J. D., Blanc, J., Truntzer, C., Desmoulins, I., Ladoire, S., Hennequin, A., Favier, L., Bengrine, L., Vincent, J., Hervieu, A., Dusserre, J. G., Lepage, C., Foucher, P., Borg, C., . . . Ghiringhelli, F. (2020). Implementation and use of whole exome sequencing for metastatic solid cancer. EBioMedicine, 51, 102624. https://doi.org/10.1016/j.ebiom.2019.102624
- Rehm, H. L., Bale, S. J., Bayrak-Toydemir, P., Berg, J. S., Brown, K. K., Deignan, J. L., Friez, M. J., Funke, B. H., Hegde, M. R., & Lyon, E. (2013). ACMG clinical laboratory standards for next-generation sequencing. Genet Med, 15(9), 733-747. https://doi.org/10.1038/gim.2013.92
- Sachdev, R., Field, M., Baynam, G. S., Beilby, J., Berarducci, M., Berman, Y., Boughtwood, T., Cusack, M. B., Fitzgerald, V., Fletcher, J., Freckmann, M. L., Grainger, N., Kirk, E., Lundie, B., Lunke, S., McGregor, L., Mowat, D., Parasivam, G., Tyrell, V., . . . A, S. L. M. (2021). Paediatric genomic testing: Navigating medicare rebatable genomic testing. J Paediatr Child Health, 57(4), 477-483. https://doi.org/10.1111/jpc.15382
- Sakai, R., Sifrim, A., Vande Moere, A., & Aerts, J. (2013). TrioVis: a visualization approach for filtering genomic variants of parent-child trios. Bioinformatics, 29(14), 1801-1802. https://doi.org/10.1093/bioinformatics/btt267
- Salfati, E. L., Spencer, E. G., Topol, S. E., Muse, E. D., Rueda, M., Lucas, J. R., Wagner, G. N., Campman, S., Topol, E. J., & Torkamani, A. (2019). Re-analysis of whole-exome sequencing data uncovers novel diagnostic variants and improves molecular diagnostic yields for sudden death and idiopathic diseases. Genome Medicine, 11(1), 83. https://doi.org/10.1186/s13073-019-0702-2
- Sanford, E. F., Clark, M. M., Farnaes, L., Williams, M. R., Perry, J. C., Ingulli, E. G., Sweeney, N. M., Doshi, A., Gold, J. J., Briggs, B., Bainbridge, M. N., Feddock, M., Watkins, K., Chowdhury, S., Nahas, S. A., Dimmock, D. P., Kingsmore, S. F., Coufal, N. G., & Investigators, R. (2019). Rapid Whole Genome Sequencing Has Clinical Utility in Children in the PICU. Pediatric critical care medicine : a journal of the Society of Critical Care Medicine and the World Federation of Pediatric Intensive and Critical Care Societies, 20(11), 1007-1020. https://doi.org/10.1097/PCC.0000000000002056
- Soden, S. E., Saunders, C. J., Willig, L. K., Farrow, E. G., Smith, L. D., Petrikin, J. E., LePichon, J. B., Miller, N. A., Thiffault, I., Dinwiddie, D. L., Twist, G., Noll, A., Heese, B. A., Zellmer, L., Atherton, A. M., Abdelmoity, A. T., Safina, N., Nyp, S. S., Zuccarelli, B., . . . Kingsmore, S. F. (2014). Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med, 6(265), 265ra168. https://doi.org/10.1126/scitranslmed.3010076
- Splinter, K., Adams, D. R., Bacino, C. A., Bellen, H. J., Bernstein, J. A., Cheatle-Jarvela, A. M., Eng, C. M., Esteves, C., Gahl, W. A., Hamid, R., Jacob, H. J., Kikani, B., Koeller, D. M., Kohane, I. S., Lee, B. H., Loscalzo, J., Luo, X., McCray, A. T., Metz, T. O., . . . Ashley, E. A. (2018). Effect of Genetic Diagnosis on Patients with Previously Undiagnosed Disease. N Engl J Med, 379(22), 2131-2139. https://doi.org/10.1056/NEJMoa1714458
- Srivastava, S., Cohen, J. S., Vernon, H., Baranano, K., McClellan, R., Jamal, L., Naidu, S., & Fatemi, A. (2014). Clinical whole exome sequencing in child neurology practice. Ann Neurol, 76(4), 473-483. https://doi.org/10.1002/ana.24251
- Tammimies, K., Marshall, C. R., Walker, S., Kaur, G., Thiruvahindrapuram, B., Lionel, A. C., Yuen, R. K., Uddin, M., Roberts, W., Weksberg, R., Woodbury-Smith, M., Zwaigenbaum, L., Anagnostou, E., Wang, Z., Wei, J., Howe, J. L., Gazzellone, M. J., Lau, L., Sung, W. W., . . . Fernandez, B. A. (2015). Molecular Diagnostic Yield of Chromosomal Microarray Analysis and Whole-Exome Sequencing in Children With Autism Spectrum Disorder. Jama, 314(9), 895-903. https://doi.org/10.1001/jama.2015.10078
- Taylor, J. C., Martin, H. C., Lise, S., Broxholme, J., Cazier, J. B., Rimmer, A., Kanapin, A., Lunter, G., Fiddy, S., Allan, C., Aricescu, A. R., Attar, M., Babbs, C., Becq, J., Beeson, D., Bento, C., Bignell, P., Blair, E., Buckle, V. J., . . . McVean, G. (2015). Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet, 47(7), 717-726. https://doi.org/10.1038/ng.3304
- Van den Veyver, I. B., Chandler, N., Wilkins-Haug, L. E., Wapner, R. J., & Chitty, L. S. (2022). International Society for Prenatal Diagnosis Updated Position Statement on the use of genome-wide sequencing for prenatal diagnosis. Prenat Diagn, 42(6), 796-803. https://doi.org/10.1002/pd.6157
- variantyx. (2020). Genomic Unity® Exome Plus Analysis. https://www.variantyx.com/products-services/rare-disorder-genetics/comprehensive-analyses/genomic-unity-exome-plus-analysis/
- Yang, Y., Muzny, D. M., Xia, F., Niu, Z., Person, R., Ding, Y., Ward, P., Braxton, A., Wang, M., Buhay, C., Veeraraghavan, N., Hawes, A., Chiang, T., Leduc, M., Beuten, J., Zhang, J., He, W., Scull, J., Willis, A., . . . Eng, C. M. (2014). Molecular findings among patients referred for clinical whole-exome sequencing. Jama, 312(18), 1870-1879. https://doi.org/10.1001/jama.2014.14601
- Yates, C. L., Monaghan, K. G., Copenheaver, D., Retterer, K., Scuffins, J., Kucera, C. R., Friedman, B., Richard, G., & Juusola, J. (2017). Whole-exome sequencing on deceased fetuses with ultrasound anomalies: expanding our knowledge of genetic disease during fetal development. Genet Med, 19(10), 1171-1178. https://doi.org/10.1038/gim.2017.31
- Zhang, J., Tang, S.-Y., Zhu, X.-B., Li, P., Lu, J.-Q., Cong, J.-S., Wang, L.-B., Zhang, F., & Li, Z. (2021). Whole exome sequencing and trio analysis to broaden the variant spectrum of genes in idiopathic hypogonadotropic hypogonadism [Original Article]. Asian Journal of Andrology, 23(3), 288-293. https://doi.org/10.4103/aja.aja_65_20
Coding Section
| CPT |
CPT Description |
| 81415 |
Exome (e.g., unexplained constitutional or heritable disorder or syndrome); sequence analysis |
| 81416 |
Exome (e.g., unexplained constitutional or heritable disorder or syndrome); sequence analysis, each comparator exome (e.g., parents, siblings) (List separately in addition to code for primary procedure) |
| 81417 |
Exome (e.g., unexplained constitutional or heritable disorder or syndrome); re-evaluation of previously obtained exome sequence (e.g., updated knowledge or unrelated condition/syndrome) |
| 81425 |
Genome (e.g., unexplained constitutional or heritable disorder or syndrome); sequence analysis |
| 81426 |
Genome (e.g., unexplained constitutional or heritable disorder or syndrome); sequence analysis, each comparator genome (e.g., parents, siblings) (List separately in addition to code for primary procedure) |
| 81427 |
Genome (e.g., unexplained constitutional or heritable disorder or syndrome); re-evaluation of previously obtained genome sequence (e.g., updated knowledge or unrelated condition/syndrome) |
| 81479 |
Unlisted molecular pathology procedure |
| 0209U |
Cytogenomic constitutional (genome-wide) analysis, interrogation of genomic regions for copy number, structural changes and areas of homozygosity for chromosomal abnormalities |
| 0214U |
Rare diseases (constitutional/heritable disorders), whole exome and mitochondrial DNA sequence analysis, including small sequence changes, deletions, duplications, short tandem repeat gene expansions, and variants in non-uniquely mappable regions, blood or saliva, identification and categorization of genetic variants, proband Proprietary test: Genomic Unity® Exome Plus Analysis -Proband, Lab/Manufacturer: Variantyx Inc, |
| 0215U |
Rare diseases (constitutional/heritable disorders), whole exome and mitochondrial DNA sequence analysis, including small sequence changes, deletions, duplications, short tandem repeat gene expansions, and variants in non-uniquely mappable regions, blood or saliva, identification and categorization of genetic variants, each comparator exome (e.g., parent, sibling) Proprietary test: Genomic Unity® Exome Plus Analysis - Comparator, Lab/Manufacturer: Variantyx Inc, Variantyx Inc |
| 0265U |
Rare constitutional and other heritable disorders, whole genome and mitochondrial DNA sequence analysis, blood, frozen and formalin-fixed paraffin-embedded (FFPE) tissue, saliva, buccal swabs or cell lines, identification of single nucleotide and copy number variants |
| 0335U |
Rare diseases (constitutional/heritable disorders), whole genome sequence analysis, including small sequence changes, copy number variants, deletions, duplications, mobile element insertions, uniparental disomy (UPD), inversions, aneuploidy, mitochondrial genome sequence analysis with heteroplasmy and large deletions, short tandem repeat (STR) gene expansions, fetal sample, identification and categorization of genetic variants |
| 0336U |
Rare diseases (constitutional/heritable disorders), whole genome sequence analysis, including small sequence changes, copy number variants, deletions, duplications, mobile element insertions, uniparental disomy (UPD), inversions, aneuploidy, mitochondrial genome sequence analysis with heteroplasmy and large deletions, short tandem repeat (STR) gene expansions, blood or saliva, identification and categorization of genetic variants, each comparator genome (e.g., parent) |
| 0425U |
Genome (e.g., unexplained constitutional or heritable disorder or syndrome), rapid sequence analysis, each comparator genome (e.g., parents, siblings) |
| 0426U |
Genome (e.g., unexplained constitutional or heritable disorder or syndrome), ultra-rapid sequence analysis |
Procedure and diagnosis codes on Medical Policy documents are included only as a general reference tool for each policy. They may not be all-inclusive.
This medical policy was developed through consideration of peer-reviewed medical literature generally recognized by the relevant medical community, U.S. FDA approval status, nationally accepted standards of medical practice and accepted standards of medical practice in this community, and other nonaffiliated technology evaluation centers, reference to federal regulations, other plan medical policies, and accredited national guidelines.
"Current Procedural Terminology © American Medical Association. All Rights Reserved"
History From 2013 Forward
| 01/28/2025 | Interim review, removing coverage criteria #8, updating wording in #4. Also updating coding. |
| 10/14/2024 | Annual review, updating policy for clarity and consistency. Coverage criteria 4 is new and addresses instances when WES is unable to identify a causative mutation. Also criteria #6 is added to address fouced eexome squencing and targeted WGS. Also updating table of terminolog, rationale, references and numerous coding updates, additions and revisions. |
| 07/29/2024 | Change review date to 10/01/2024. |
| 07/26/2023 | Annual review, no change to policy intent. Policy updated for clarity and consistency. Also updating description, rationale and references. |
| 11/07/2022 | Annual review moved to October. Updating rationale and references, no other changes. |
| 08/01/2022 | Annual review, no change to policy intent. Updating coding, description, rationale and references |
| 06/15/2022 | Added code 0329U |
| 07/13/2021 | Annual review, no change to policy intent. Updating coding, rationale and references. |
| 07/01/2020 | Annual review, adding two new areas of coverage criteria, fetus with ultrasound abnormalities and comparator analysis with specific criteria for each. Reformatting policy for clarity. |
| 07/12/2019 | Annual review, no change to policy intent, updating title and including the acronym for Whole Exome Sequencing in the policy, also updating coding.. |
| 07/18/2018 | Annual review, no change to policy intent. |
| 11/01/2017 | Interim review, updating policy verbiage with medical necessity criteria for services previously considered investigational for all indications. Also updating background, description, regulatory status, rationale and references. |
| 07/19/2017 | Annual review, no change to policy intent. |
| 04/25/2017 | Updated category to Laboratory. No other changes. |
| 10/11/2016 | Annual review, no change to policy intent. |
| 12/1/2016 | Update CPT codes with 2016 codes. No change in policy intent. |
| 10/27/2015 | Annual review, no change to policy intent. Updating background, description, rationale and references. |
| 10/20/2014 | Annual review, extensive revision as policy now addresses whole genome sequencing as well as whole exome sequencing. Added coding & policy guidelines. Updated entire policy. |
| 03/6/2014 | Corrected Last Reviewed Date |
| 10/16/2013 | NEW POLICY |